Презентація на тему:
Клінічний розбір хворих з лейкеміями
Завантажити презентацію


- пухлина органів кровотворення Гострі лейкемії...")








")

— злоякісне захворювання крові, що характери...")
: Доб...")
")
 лімфоаденопатія- наявність збільшених ...")
...")











, я...")

 Хвороба Рустицького-Калера O.Kahler (...")


- чоловіки- 9,9 жінки...")

 серед хворих моноклональними гамапатіями складаю...")


 особливості: немає нефротичного синдрому, асциту, ...")








Клінічний розбір хворих з лейкеміями
Завантажити презентаціюПрезентація по слайдам:

Клінічний розбір хворих з лейкеміями Лектор: Лихацька В.О.
- пухлина органів кровотворення Гострі лейкемії...")
Лейкемія (лейкоз, гемобластоз)- пухлина органів кровотворення Гострі лейкемії Хронічні лейкемії: а) мієлопроліферативні; б) лімфопроліферативні

онковіруси Ендо- екзоканцерогени радіація Соматична мутація Порушення проліферації і диференціювання Накопичення клітин Пригнічення кровотвотвореня Прогресія пухлинного клону Метастазування в органи і тканини Анемія, Геморагії, Імунодефіцит, Нейролейкемія, інтоксикація Розлад регуляції, резистентність до лікування

Класифікація стадій гострих лейкозів 1. Початкова стадія – виявити вчасно досить важко. 2. Розгорнутий період захворювання- характеризується значним пригніченням нормального кровотворення, високим бластозом у кістковому мозку. 3. Повна ремісія - це стан, при якому в пунктаті кісткового мозку знаходять не більше 5% бластних клітин, або загальна кількість лімфоїдних клітин менш ніж 40%. Екстрамедулярні лейкозні проліферати відсутні. 4. Одужанням від гострого лейкозу вважається повна ремісія протягом 5 років (безрецидивне 5-річне виживання). 5. Рецидив ГЛ може бути кістково-мозковим або місцевим. 6. Термінальна стадія - поняття умовне, відображає сучасні терапевтичні можливості і інкурабельний стан пухлинної прогресії.

Класифікація лейкемій ГМЛ: Гострий мієлоїдний лейкоз з мінімальними ознаками диференціювання - ГМЛ Мо. Гострий мієлоїдний лейкоз без ознак визрівання- ГМЛ МІ. Гострий мієлоїдний лейкоз з ознаками визрівання- ГМЛ М2. Гострий промієлоцитарний лейкоз - ГМЛ МЗ. Гострий мієломоноцитарний лейкоз - ГМЛ М4. Гострий моноцитарний лейкоз - ГМЛ М5. Еритролейкоз - ГМЛ М6. Гострий мегакаріобластний лейкоз - ГМЛ М7. Сучасна FАВ-класифікаційна система описує 3 підтипи ГЛЛ (L1,L2,L3), які визначаються за індивідуальними цитологічними ознаками, такими, як розмір клітин, характер ядерного хроматину, форма ядра, ядерець та інтенсивність базофілії цитоплазми

Імунобіологія гострих лейкозів У відповідності з пропозиціями першої Європейської групи по імунологічній класифікації лейкозів виділяють основні 4 форми ГЛ та ряд їх субтипів. 1 Гострі лімфобластні лейкози (ГЛЛ): В-лінійні: В-І (про-В) ГЛЛ В-ІІ (соттоп) ГЛЛ В-ПІ (пре-В) ГЛЛ В-ІУ (зрілий-В) ГЛЛ Т-лінійні: Т-І (про-Т) ГЛЛ Т-П (пре-Т) ГЛЛ Т-ІП (кортикальний) ГЛЛ Т-ІУ (зрілий-Т) ГЛЛ Т-о/ріу/5 ГЛЛ з експресією 1 або 2 мієлоїдних маркерів (мієло+ГЛЛ) 2 Гострі мієлоїдні лейкози (ГМЛ) - мієломоноцитарний - еритроїдний (ранній) незрілий і зрілий - мегакаріоцитарний - низькодиференційований мієлоїдний (ГМЛ-МО) - Т(іТ+ГМЛ - ГМЛ з експресією 1 або 2 лімфоїдних маркерів (лімф.+ГМЛ) 3. Біфенотипічні гострі лейкози 4.Недиферещійовані гострі лейкози

Критерії діагностики гострої лейкемії Клінічні: ГЛ характеризується наявністю: непластичного синдрому (лімфопроліферативний, мієлопроліферативного)- збільшенних периферичних лімфатичних вузлів, печінки та селезінки, виразково-некротичного синдрому, анемічного, геморагічного інтоксикаційного синдромів, наявністю осалгій, стерналгій, артралгій, імунодефіциту, септичних ускладнень, нейролейкозу

Лабораторні критерії ГЛ Критерієм постановки діагнозу гострої лейкемії є наявність більше 30% бластних клітин у кістковому мозку

Таблиця 1. Цитохімічна характеристика форм лейкозів

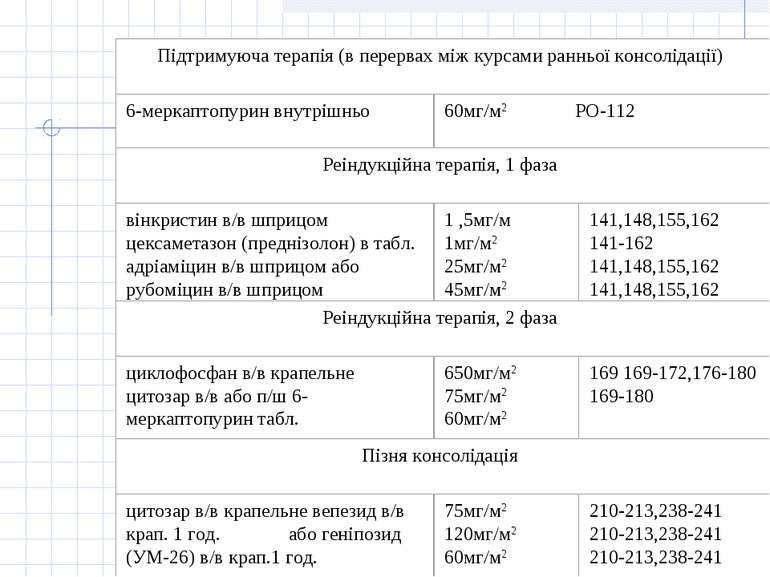
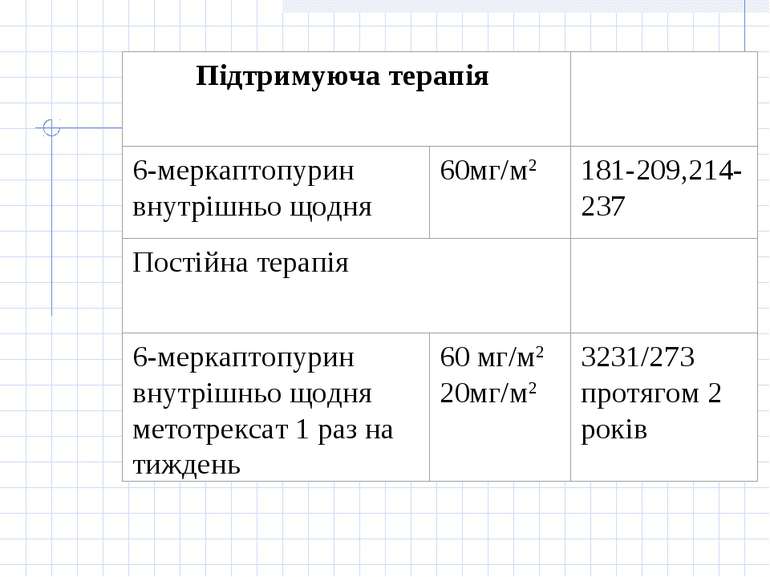
Принцип лікування гострої лімфобластної лейкемії полягає у застосуванні наступних етапів: 1.Індукція ремісії. Метою фази індукції ремісії є знищення клону лейкемічних клітин. 2. Консолідація/інтенсифікація ремісії. Метою фази консолідації/інтенсифікації ремісії є ліквідація "залишкової хвороби", що досягається застосуванням високих доз цитостатиків (цитозару та метотрексату), а також поєднання циклофосфану з середніми дозами цитозару або етопозиду з цитозаром. Найвагомішою побічною дією при цьому є мієлосупресія. 3. Підтримуюче лікування. Метою підтримуючого лікування є запобігання рецидиву захворювання і полягає у тривалому застосуванні „легкої" хіміотерапії без вираженої мієлосупресії або поєднання її з ротаційними курсами полі хіміотерапії з високими дозами цитостатиків. У хворих з високої групи ризику та у хворих з рецидивом захворювання у період ремісії після консолідації ремісії показана мієлотрансплантація. 4. Профілактика нейролейкемії
")
Група "стандартного ризику" Протокол ПХТ(В.НоеІzег еt аl., 1988р.)
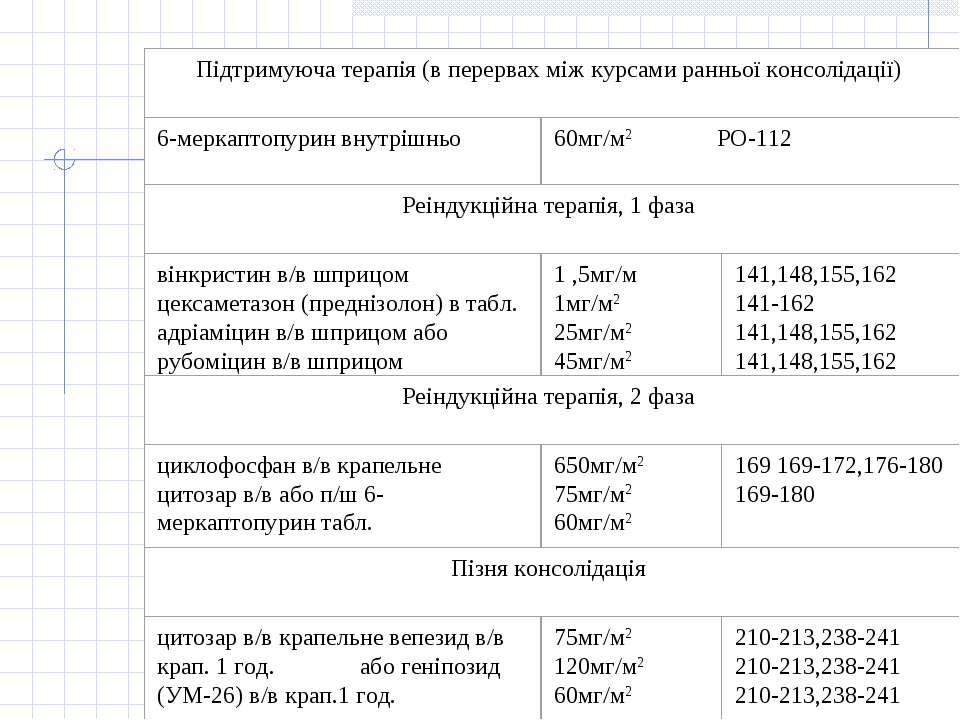
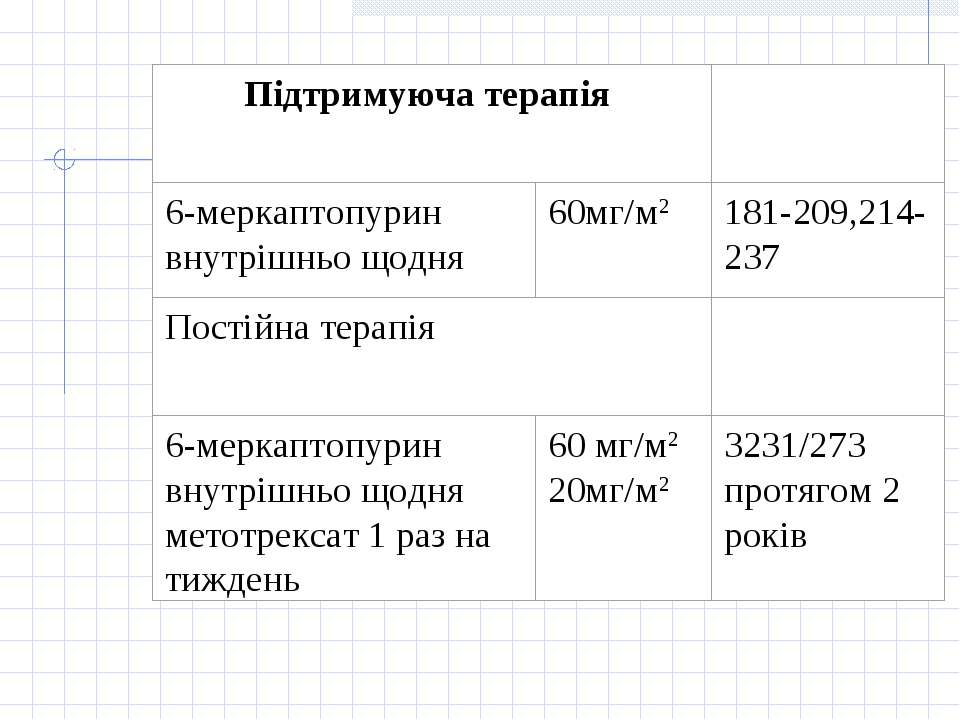

Схема ПХТ при ГМЛ
— злоякісне захворювання крові, що характери...")
Хронічна лімфоїдна лейкемія (ХЛЛ)— злоякісне захворювання крові, що характеризується акумуляцією непроліферуючих морфологічно зрілих лімфоїдних клітин у периферичній крові, кістковому мозку, лімфатичних вузлах та селезінці.
: Доб...")
Хронічна лімфоцитарна лейкемія КЛІНІЧНІ ФОРМИ ХЛЛ (А.І. Воробйов, 1999): Доброякісна Прогресуюча Спленомегалічна. Абдомінальна. Пухлинна. Кістковомозкова. Пролімфоцитарна. Т-клітинна. КЛІНІЧНА КАРТИНА: Синдроми: - неопластичний; - поліаденопатія; - спленомегалія; - гепатомегалія; розлади імунітету. ЗА ПЕРЕБІГОМ: - початковий період; - період виражених клінічних проявів; термінальна стадія.
")
Система клінічних стадій ХЛЛ (Rai Staging System, 1990)
 лімфоаденопатія- наявність збільшених ...")
5. Діагностичні критерії: Клінічні: а) лімфоаденопатія- наявність збільшених лімфовузлів тістовидної консистенції, не спаяних з оточуючими тканинами, не болючих, які рухаються при пальпації; б) гепатомегалія, спленомегалія; В більш пізніх стадіях хвороби приєднуються анемія, тромбоцитопенія та імунодефіцит. Лабораторні критерії діагностики: 1) абсолютний лімфоцитоз у периферичній крові (>5 Г/л), у мазку крові домінують зрілі малі лімфоцити, відсоток їх попередників (лімфобластів та пролімфоцитів) не перевищує 10; 2) у кістковому мозку > 40 % лімфоцитів; 4) низька щільність поверхневих імуноглобулінів; 5) імунологічний фенотип лейкемічних В-лімфоцитів: СDІ9+ СD20+СD22-СD24+СD25-СD23+,
...")
КРИТЕРІЇ ДІАГНОЗУ ХЛЛ (запропоновані Міжнародною робочою нарадою з ХЛЛ, 1989): абсолютна кількість лімфоцитів у периферичній крові > 10 · 10 9/л; переважно зрілих лімфоцитів; кількість лімфоцитів у кістковому мозку > 30 % всіх ядровмісних клітин; лімфоцити мають імунологічні маркери, які підтверджують їх належність до В-клітинного клону лейкозних клітин. Діагноз ставиться при наявності 3 критеріїв, коли лімфоцитів менше 10 · 10 9/л. діагноз ХЛЛ ставиться за наявності другого і третього критерію.

ДИФЕРЕНЦІЙНА ДІАГНОСТИКА Доброякісні лімфоцитози: при мононуклеозі; бактеріальні і вірус інфукціях при імунних захворюваннях; при спленектомії; лімфоцитоз у курців Злоякісні гемобластози: гострі лейкемії; лейкемія з лімфомних клітин; лімфомамантійної зони; волосатоклітинна лейкемія; селезінкова лімфома з ворсинчастими клітинами; макроглобулінемія Вальденстрема; Т-клітинні лейкемії

ПРИНЦИПИ ЛІКУВАННЯ Лікувальні підходи • Лише спостереження. Пацієнта оглядають кожних 3-6 місяців, а лікування розпочинають у випадках появи неопластичного синдрому або цитопеніі (анемія або тромбоцитопенія). • Хлорамбуцил залишається "стандартним" лікувальним засобом. Його застосову ють або тривало, або переривчастими циклами (щомісяця упродовж 5-ти днів по 0,3 мг/кг/день, часто в поєднанні з кортикостероїдами). Лікування припиняють у випадку досягнення максимальної регресії і поновлюють при рецидиві. • "Агресивна" хіміотерапія базується на 3-х групах медикаментів: • циклофосфамід застосовують часто внутрішньовенне, наприклад, у складі схеми СОР: циклофосфамід, вінкристин, кортикостероїди. Порівняно з терапією хлорамбуцилом названа схема дає більше ускладнень без доведеної вищої ефективності; • антрацикліни, зокрема доксорубіцин, входять до складу схеми тіга-СНОР: доксорубіцин та вінкристин внутрішньовенне на 1-й день, циклофосфамід та кортикостероїди рег os з 1-го по 5-й день. Названа схема ефективна у випадках ХЛЛ з цитопенією або неопластичним синдромом; • Інтерферон-а при ХЛЛ застосовують лише в поодиноких випадках.

Аналоги пуринів. Флюдарабін вводять упродовж 5-ти днів щомісяця. Цей медикамент не має типової токсичності, характерної для цитостатиків (нудота, блювання, алопеція і т. д.), але призводить до тривалої цитопеніі та імунодепресії з інфекційними (пневмонія, герпес і т. д.) та імунологічними (автоімунна гемолітична анемія) ускладненнями. Безпосередня ефективність флюдарабіну є високою, особливо в раніше не лікованих пацієнтів. Кількість повних регресій сягає значних величин, вра ховуючи повну медулярну регресію, іноді з повним "зникненням" злояхісного клону. • Пересадка кісткового мозку. Застосовують алогенну трансплантацію від членів родини та пересадку кісткового мозку або клітин-попереднихів. Підготовче лікування базується, як звичайно, на поєднанні циклофосфаміду з тотальним опроміненням тіла пацієнта. • Спленектомію проводять для зменшення маси пухлини, корекції гіперспленізму або резистентної до кортикостероїдів імунної цитопенії. • Променеву терапію проводять як паліативний захід у випадках збільшення лімфатичних вузлів, неоперабельної спленомегалії з больовим синдромом. Гемікорпоральне опромінення в комбінації з факторами росту в пацієнтів, резистентних до хіміотерапії, перебуває у фазі клінічних дослідженнь

ХМЛ –пухлинне захворювання крові, яке характеризується моноклональною проліферацією на рівні плюрипотентних гематопоетичних стовбурових клітин, яка призводить до гіперплазії клітин гранулоцитарного ряду. Класифікація Клінічні варіанти ХМЛ: типовий ХМЛ Ph+) атиповий ХМЛ (Ph-) ХМЛ у дітей МОРФОЛОГІЧНІ ВАРІАНТИ ХМЛ: Хронічна еозинофільна лейкемія; Хронічна базофільна лейкемія; Хронічна моноцитарна лейкемія; Хронічна нейтрофільна лейкемія. ФАЗИ КЛІНІЧНОГО ПЕРЕБІГУ: хронічна прогресуюча (акселерації) гостра (бластний криз Стадії: Початкова; Рознорнута; термінальна

Епідеміологія На Заході щороку діагностують 10 випадків ХМЛ на мільйон населеня. У Франції виявляють 600 нових випадків захворювання на рік. Частота виявлення ХМЛ є в 4-5 разів меншою, ніж частота гострих лейкозів. Вона прогресивно зростає з віком - від одного випадку на мільйон дитячого населення-віком до 10 років до більш як 30 випадків на мільйон дорослого населення віком понад 60 років. Згідно з даними різних досліджень, середній вік на момент встановлення діагнозу коливається від 30-ти до 60-ти років. ХМЛ дещо частіше трапляється в чоловіків, ніж у жінок.

Етіологія і патогенез Філадельфійська хромосома - це результат реципрокної транслокації між 9-ю та 22-ю хромосомами, пошкодженими відповідно в ділянках д34 та q11. Матрична РНК сприяє утворенню протеїну, який завдяки тирозинкіназній активності перетворює нормальні гематопоетичні клітини в лейкемічні.

Діагностичні критерії: -Клінічні- мієлопроліферативний синдром, який проявляється спленомегалією, гепатомегалією, лейкоцитозом з проліферацією клітин гранулоцитарного ряду. При прогресуванні хвороби наростає анемія, тромбоцитопенія і в фазі бластого кризу ХМЛ трансформується у гостру лейкемію з відповідною клінічною симптоматикою (анемічний та геморагічні синдроми, нейролейкоз, септичні ускладнення, та ін.). Хронічна мієлоїдна лейкемія - це класичне гематологічне захворювання, яке проходить три стадії розвитку: хронічну стадію, стадію акселерації та стадію трансформації в гостру лейкемію. Гранулоцитарний нейтрофільний гіперлейкоцитоз з мієлеміею повинен насторожити лікаря. Підозру на ХМЛ підтверджують еозинофілія, базофілія та гіпертромбоцитемія.

Критерії діагностики хронічної фази ХМЛ Периферична кров: -лейкоцитоз > 30,0 Г/л зі зсувом у лейкограмі; -морфологія гранулоцитарних клітин нормальна, диспластичних гранулоцитів < 10 % ; -базофільно-еозинофільна асоціація (збільшення частки базофілів та еозинофілів); -зниження до повної відсутності активності лужної фосфатази нейтрофілів (ПАЛФН 10; -наявність Ph+- хромосоми в клітинах мієло - та лімфопоезу

Критерії діагностики фази акселерації ХМЛ: -зростання кількості лейкоцитів, незважаючи на лікування, яке проводиться; гарячка понад 38,5°С протягом тижня без ознак інфекції; -втрата ваги понад 2 кг протягом ЗО днів; - зниження кількості тромбоцитів понад 50 % вихідного рівня протягом ЗО днів; - прогредієнтна тромбоцитопенія нижче 100,0 Г/л; - прогредієнтне зниження рівня гемоглобіну нижче 90 г/л; - прогредієнтне збільшення селезінки; - збільшення кількості бластів більше 10 % у периферичній крові або кістковому мозку; - збільшення базофілів у периферичній крові > 20 %; - поява додаткових цитогенетичних аномалій (подвоєння Ph+ 8.+19, -7

Критерії діагностики бластної кризи ХМЛ: кількість бластів і промієлоцитів периферичної крові >30 %, кількість бластів і промієлоцитів у кістковому мозку >50 % всіх ядерних елементів, цитологічне або гістологічне підтверджені екстрамедулярні бластні інфільтрати.

Лікувальна тактика при ХМЛ полягає: у проведенні паліативної терапії, яка дозволяє контролювати мієлоїдну проліферацію (гідроксисечовина, бусульфан, мієлосан); проведення ерадикації лейкемічного клону, кінцевою метою якої є одужання пацієнта (алотрансплантація кісткового мозку, автотрансплантація кісткового мозку або стовбурових гемопоетичних клітин, іматініб (Глівек), препарати а-інтерферону (Інтрон А, Реальдирон, Роферон-А). Вибір лікування залежить від стадії захворювання, віку пацієнта, прогностичнихпоказників, наявності гістосумісного родинного донора
, я...")
У закордонних клініках майже не застосовують бусульфан (мілєран, мієлосан), який в Україні є популярним у зв'язку з доведеним цитостатичним ефектом і невисокою вартістю лікування. Доза бусульфану призначається в залежності від числа лейкоцитів. При кількості лейкоцитів 150-200 Г/л призначають 6-8 мг бусульфану на добу. Коли кількість лейкоцитів знизиться до 100 Г/л, дозу препарата знижують до 4-6 мг/добу, а при зниженні кількості лейкоцитів до 40-60 Г/л добову дозу бусульфана зменшують до 2-4 мг. При зниженні числа лейкоцитів до 20-10 Г/л переходять на підтримуючу терапію (2 мг один раз в 7-10 днів). Підтримуючу дозу бусульфана індивідуально підбирають так, щоб число лейкоцитів утримувати на рівні 8-12 Г/л. Не можна починати лікування з бусульфану у хворих, яким планується проведення трансплантації кісткового мозку або стовбурових гемопоетичних клітин.

Після зниження рівня лейкоцитів до нормальних цифр хворих з групи високого ризику та молодих пацієнтів готують до алотрансплантації. При відсутності донора продовжують лікування гідроксисечовиною або проводять терапію іматінібом (Глівек), -інтерфероном (Інтрон А, Реальдирон, Роферон-А) з наступним розглядом варіанту проведення автотрансплантації кісткового мозку. При відсутності донора і неможливості проведення автотрансплантації продовжують лікування препаратами -інтерферону. Оптимальна добова доза -інтерферону призначається з розрахунку 5 млн/м2 щоденно.
 Хвороба Рустицького-Калера O.Kahler (...")
Мієломна хвороба (Г.А.Алексеев- 1949р.) Хвороба Рустицького-Калера O.Kahler (1889р) Множинна мієлома - це моноклональний пухлинний процес з атипових плазмоцитів, розвиток якого пов'язаний з проліферацією та накопиченням імуноглобулін - секретуючих В-клітин.

Мієломна хвороба- це лімфопроліферативне захворювання, морфологічним субстратом якого є плазматичні клітини, що продукують патологічний імуноглобулін

Мієломна хвороба складає: 1% онкологічних захворювань; 10% всіх гемобластозів; Хворіють рідше люди жовтої раси; Частіше- негри.
- чоловіки- 9,9 жінки...")
Поширеність: Китай- 1.0 на 100000 населення; США (негри)- чоловіки- 9,9 жінки-6,7 Європа: Англія- 2,6 на 100000 населення, Швеція- 3.3 на 100000 населення

Мієломна хвороба- Захворювання людей, старших за 40 років, середній вік- 69 років. Молоді до 30 років- рідкість
 серед хворих моноклональними гамапатіями складаю...")
За даними клініки Мейо (США) серед хворих моноклональними гамапатіями складають: 52% хворих неясного генезу; 12%-хворі на амілоїдоз; 33%-онкологічні хворі; 19%-мієломна хвороба.

Клінічна симптоматика: Синдром кісткової патології (оссалгії, пухлиноподібні - деформації кісток, компресійні переломи, “дірявий череп”, “симптом пробійника”, “бджолині соти”, “риб ячі хребці”, остеопороз).

Синдром білкової патології (гіперпротеїнемія, нормального -глобуліну, наявність М-градієнта при електрофорезі білків, стійка протеїнурія, розвиток амілоїдозу.
 особливості: немає нефротичного синдрому, асциту, ...")
Мієломна нефропатія- (ХНН) особливості: немає нефротичного синдрому, асциту, гіперхолестеринемії, гіпертензії, альбуміни-нормальні, ).

Клінічні синдроми Вісцеральної патології- Гепатомегалія Спленомегалія Ураження системи кровотворення, Анемія; Панцитопенія; Прискорена ШЗЕ Підвищеної в язкості- порушення зору, порушення периферичного кровообігу. Неврологічні прояви- результат компресії і інфільтрації.

Клінічні синдроми інфекційні ускладненя Гіперкальціємічний синдром - остеолізис, нефрокальциноз

1. Анамнез: болі в кістках, радикулярні болі, зменшення росту, кровоточивість слизових, часті бактеріальні інфекції. 2. Огляд хворого: звернути увагу на колір шкіри та слизових, наявність геморагічного синдрому, зміни з боку опорно-рухового апарату (переломи, болючі утвори на кістках), зміни зі сторони нирок. 3. Лабораторне обстеження: загальний аналіз крові з визначенням кількості тромбоцитів, загальний аналіз сечі. Біохімічні дослідження: загальний білок сироватки крові, рівень креатиніну, сечовини,кальцію. 4. Інструментальне обстеження: флюорографія органів грудної клітки, рентгенографія хребта, черепа, здухвинних та інших кісток. Сонографічне дослідження. Особливості обстеження:

Діагностичні критерії мієломної хвороби Основні: 1. Плазматична інфільтрація кісткового мозку (кількість плазматичних клітин більше 15 %) 2. Моноклональна Ig-патія (сироватковий М-компонент та /або білок Бенс-Джонса у сечі) 3. Ураження плоских кісток та хребта (остеодеструкція, остеопороз) Допоміжні 1.Стійке та тривале збільшення ШОЕ 2. Різко виражене збільшення вмісту загального білка у сироватці крові 3. Гіперкальціемія 4. Гіпогаммаглобулінемія 5. Наростаюча анемія

СТАДІЇ МНОЖИННОЇ МІЄЛОМИ Перша стадія: Сукупність наступних ознак: рівень гемоглобіну більше 100г/л, гематокрит більше 32 % рівень Са у сироватці крові менше 3ммоль/л відсутність остеолізу або солітарне кісткове вогнище низький рівень М-компонента а) IgG менше 50 г/л б) IgA менше 30 г/л в) легкі ланцюги у сечі менше 4 г/добу

СТАДІЇ МНОЖИННОЇ МІЄЛОМИ Друга стадія: Сукупність наступних ознак: рівень гемоглобіну 85-100 г/л, гематокрит 25-32% рівень Са у сироватці крові 3ммоль/л наявність остеолізу рівень М-компонента а) IgG менше 50 г/л 50-70 г/л б) IgA менше 30 г/л 30-50 г/л в) легкі ланцюги у сечі менше 4-12 г/добу

СТАДІЇ МНОЖИННОЇ МІЄЛОМИ Третя стадія: Наявність одного або більше із наступних ознак: рівень гемоглобіну менше 85 г/л, гематокрит менше 25% рівень Са у сироватці крові більше 3ммоль/л виражений остеоліз високий рівень продукції М-компонента а) IgG більше 70 г/л б) IgA більше 50 г/л в) легкі ланцюги у сечі більше 12 г/добу

СХЕМА ПОЛІХІМІОТЕРАПІЇ МНОЖИННОЇ МІЄЛОМИ.
Схожі презентації











Категорії