Презентація на тему:
Предмет і завдання патологічної фізіології.Загальне вчення про хворобу, етіологію та патогенез.Роль спадковості в патології людини
Завантажити презентацію











 — це вчення про причини...")
 — це вчення п...")
 РОЗВИТКУ ХВОРОБИ")
 триває в...")


, рецидив, перехід...")











 можуть бути викликані різними причинами...")

 і анафазного відставання (Б) хромосом")

 називають такі, коли кількіст...")































, якому вист...")


 запобігання впливу фізичних та хімічних мутагенів, н...")









Предмет і завдання патологічної фізіології.Загальне вчення про хворобу, етіологію та патогенез.Роль спадковості в патології людини
Завантажити презентаціюПрезентація по слайдам:

Предмет і завдання патологічної фізіології. Загальне вчення про хворобу, етіологію та патогенез. Роль спадковості в патології людини

ПРЕДМЕТ І ЗАВДАННЯ ПАТОЛОГІЧНОЇ ФІЗІОЛОГІЇ Патологічна фізіологія — це наука, що вивчає життєдіяльність хворого організму. Патологічна фізіологія вивчає основні закономірності виникнення, розвитку і кінця хвороби.

Курс патологічної фізіології поділяється на три частини. Перша частина — нозологія Друга частина — типові патологічні процеси Третя частина — спеціальна патологічна фізіологія.

ЕКСПЕРИМЕНТ ЯК ОСНОВНИЙ МЕТОД ПАТОЛОГІЧНОЇ ФІЗІОЛОГІЇ Об'єктом вивчення патологічної фізіології є хвороба Методом — патофізіологічний експеримент.

ЗВ'ЯЗОК ПАТОЛОГІЧНОЇ ФІЗІОЛОГІЇ 3 ІНШИМИ МЕДИЧНИМИ НАУКАМИ, ЇЇ ЗНАЧЕННЯ ДЛЯ КЛІНІКИ

ЗАГАЛЬНЕ ВЧЕННЯ ПРО ХВОРОБУ Вчення про хворобу, або загальна нозологія (від грец. поsоs— хвороба)

ВООЗ: «Здоров'я—це стан повного фізичного, психологічного й соціального благополуччя, а не тільки відсутність хвороб або фізичних вад»

Здоров'я— це насамперед стан організму, в якому спостерігаються відповідність структури і функції, а також здатність регуляторних систем підтримувати сталість внутрішнього середовища (гомеостаз)

Хвороба — це порушення нормальної життєдіяльності організму внаслідок впливу на нього шкідливих агентів, що спричинює зниження його пристосовних можливостей, працездатності й збільшує можливість смерті.

ВЧЕННЯ ПРО ЕТІОЛОГІЮ І ПАТОГЕНЕЗ
 — це вчення про причини...")
Етіологія (від грец. аіtіа — причина, lоgоs — вчення) — це вчення про причини й умови виникнення хвороби.
 — це вчення п...")
Патогенез (від грец. pathos — страждання, genesis — походження) — це вчення про механізми розвитку і кінця хвороби.
 РОЗВИТКУ ХВОРОБИ")
ОСНОВНІ ПЕРІОДИ (стадії) РОЗВИТКУ ХВОРОБИ
 триває в...")
Латентний період (стосовно інфекційних хвороб — інкубаційний період) триває від моменту впливу причини до появи перших клінічних ознак хвороби.

Продромальний період — відрізок часу від перших ознак хвороби до повного її прояву.

Період виражених проявів, або розпалу, хвороби характеризується повним розвитком клінічної картини
, рецидив, перехід...")
Кінець хвороби може бути такий: видужання (повне й неповне), рецидив, перехід у хронічну форму, смерть.

Видужання— це процес, який приводить до ліквідації порушень, спричинених хворобою, і відновлення нормальних відношень організму із середовищем, для людини — насамперед до відновлення її працездатності.

Рецидив — це новий прояв хвороби після удаваного або неповного її припинення

Перехід у хронічну форму означає, що хвороба перебігає повільно з тривалими періодами ремісії (місяці й навіть роки).

Спадковість - це здатність живих організмів відтворювати собі подібних, іншими словами, це властивість живої особини передавати нащадкам притаманний їй тип обміну речовин.

Структурна одиниця спадковості - ген. Він являє собою ділянку молекули ДНК із специфічною послідовністю пуринових і пірамідинових основ, яка відповідає за біосинтез певного білка. В клітинному ядрі гени разом з білками, ферментами і РНК упаковані в особливі структури, названі хромосомами.

У здорової людини в соматичних клітинах нараховується 46 хромосом, тобто 23 пари, в тому числі 22 пари аутосом і одна пара статевих хромосом. Це так званий диплоїдний (подвійний) набір. У статевих клітинах (сперматоцитах і ооцитах) міститься одинарна кількість хромосом - 23 (гаплоїдний набір).


Головна властивість генів - їх здатність передаватися з покоління в покоління в незміненому вигляді. Гени як носії спадкової інформації повинні бути здатними до випадкових змін. Такі зміни дійсно існують, і їх називають мутаціями.

Мутації можуть виникати і в статевих, і в соматичних клітинах. Ті, що виникають у статевих клітинах, передаються особинам наступного покоління і виявляються в клітинах нащадків, які стали їх носіями. Соматичні мутації можна виявити лише в потомстві відповідної мутантної клітини і то лише за умови, що вони перешкоджають клітині здійснювати її специфічні функції, тобто проявляються фенотипічно.

Розрізняють три типи мутацій - геномні хромосомні генні

Геномні мутації полягають у зміні кількості хромосом. Можливі два варіанти: а) анеуплоїдія - дефіцит або надлишок окремих хромосом (моносомія, трисомія) б) поліплоїдія, тобто кратне збільшення генома
 можуть бути викликані різними причинами...")
Аномалії числа хромосом (анеуплоїдії) можуть бути викликані різними причинами. Найважливішим механізмом їх появи вважається нерозходження хромосом під час клітинного поділу.

Наступним механізмом, який обумовлює геномні мутації, є втрата окремої хромосоми внаслідок так званого анафазного відставання, коли одна із хромосом відстає від інших під час руху до полюса і безслідно гине.
 і анафазного відставання (Б) хромосом")
Схематичне зображення нерозходження (А) і анафазного відставання (Б) хромосом

Третім механізмом геномних мутацій виступає поліплоїдизація, коли в кожній клітині геном збільшується у 2-3 рази. Максимальна кількість хромосом, що зареєстрована у людини - 69 (триплоїдія).
 називають такі, коли кількіст...")
Хромосомними мутаціями (аномаліями, абераціями) називають такі, коли кількість хромосом не змінюється, зате порушується їх структура.

Найчастіші з них: делеція - випадіння ділянки хромосоми; інверсія - коли фрагмент хромосоми перевертається на 1800 ; кільцювання - коли відриваються обидва кінці хромосоми і вона згортається в кільце; транслокація - обмін генетичного матеріалу між хромосомами. Геномні і хромосомні аномалії вивчають під мікроскопом.

Генні мутації не можна виявити за допомогою мікроскопа, а лише шляхом генетичного аналізу фенотипічних змін. Суть їх у тому, що в ДНК порушується послідовність пуринових і піримідинових основ, тобто порушується хімічна будова генів. Якщо мутація сталася в статевій клітині, то дефектний ген передається особинам наступних поколінь. Так виникають спадкові хвороби.

Фактори зовнішнього середовища, здатні викликати мутації, називаються мутагенами.

Вони поділяються на три групи: фізичні, хімічні біологічні

До найсильніших фізичних мутагенів належать іонізуючі промені. Вони викликають переважно хромосомні і геномні мутації. Серед хімічних мутагенів найбільшою активністю відзначаються азотистий іприт, ефіри метилсульфонових кислот, гідроксиламін, акридинові барвники (трипафлавін), аналоги азотистих основ (5-бромурацил). Усі вони порушують будову ДНК. До біологічних мутагенів відносять віруси кору, корової краснухи, гепатиту.

Хвороби, які виникають на грунті пошкодження генетичного апарату, називають спадковими. Насправді не всі вони спадкуються. Їх носії або рано помирають, або неспроможні мати потомство. Зараз зареєстровано понад 2000 спадкових хвороб.

Спадкові хвороби поділяються на дві групи: хромосомні (результат геномних і хромосомних мутацій) генні (результат генних мутацій).

Хромосомних хвороб відомо біля 300.

Хвороба Дауна відома з 1866 р. як різновид розумової відсталості. Розрізняють три генетичних варіанти хвороби - класичний, транслокаційний і на грунті мозаїцизму. Класичний варіант зумовлений каріотипом 47,+21, тобто це трисомія за 21 парою. Такий каріотип утворюється під час поділу статевих клітин, якщо хромосоми 21 пари не розходяться і в анафазі відходять ло одного й того ж полюса.

Такі хворі низькі на зріст, у них широке плоске лице, широке перенісся, косий розріз очей (монголізм), напіввідкритий рот, прирослі мочки вух, плоска потилиця. Кінцівки короткі, пальці часто зрощені, на долонях одна поперечна лінія (“мавп’яча”) замість однієї поздовжньої і двох поперечних. Частота хвороби зростає з віком батьків. В сім’ї реєструється, як правило, один хворий, повторні випадки рідкі. Тривалість життя хворих вкорочена. Третина їх помирає до кінця першого року життя, ще половина - до кінця третього року, а ті, що вижили, старіють раніше, ніж здорові люди. Хворі з класичною хворобою Дауна безплідні, тому вони не передаються по спадковості. Частота класичного варіанту - 1-2/1000.

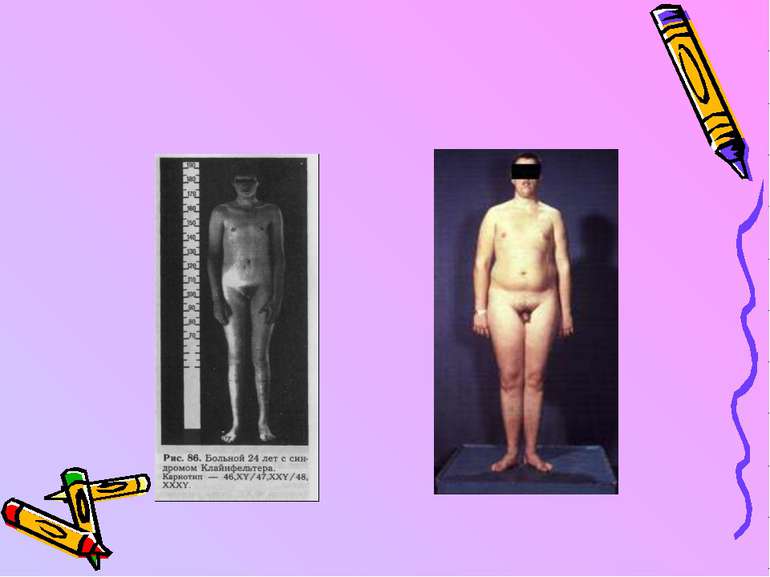
Синдром Кляйнфельтера описаний як гіпогонадизм у чоловіка. Частота його - 1/700. Хворі з класичним варіантом мають каріотип 47,ХХY. Причина його виникнення - нерозходження Х-хромосом під час поділу статевих клітин. Пізніше були знайдені каріотипи з більшою кількістю Х-хромосом (ХХХY, ХХХХY). Чим більше зайвих Х-хромосом, тим важча клінічна картина.

Karyotype of Klinefelter Syndrome - 47,XXY

Характерні ознаки синдрому - високий зріст, гінекомастія, атрофія яєчок, жіночий тип оволосіння лобка, високий голос, безпліддя, остеопороз, розумова відсталість. Частота синдрому зростає з віком матері, але не залежить від віку батька. Хворі не мають потомства. Частіше все таки трапляється не класичний варіант, а мозаїцизм 46,XY/47,XXY. Цим можна пояснити стерті і рудиментарні форми синдрому.
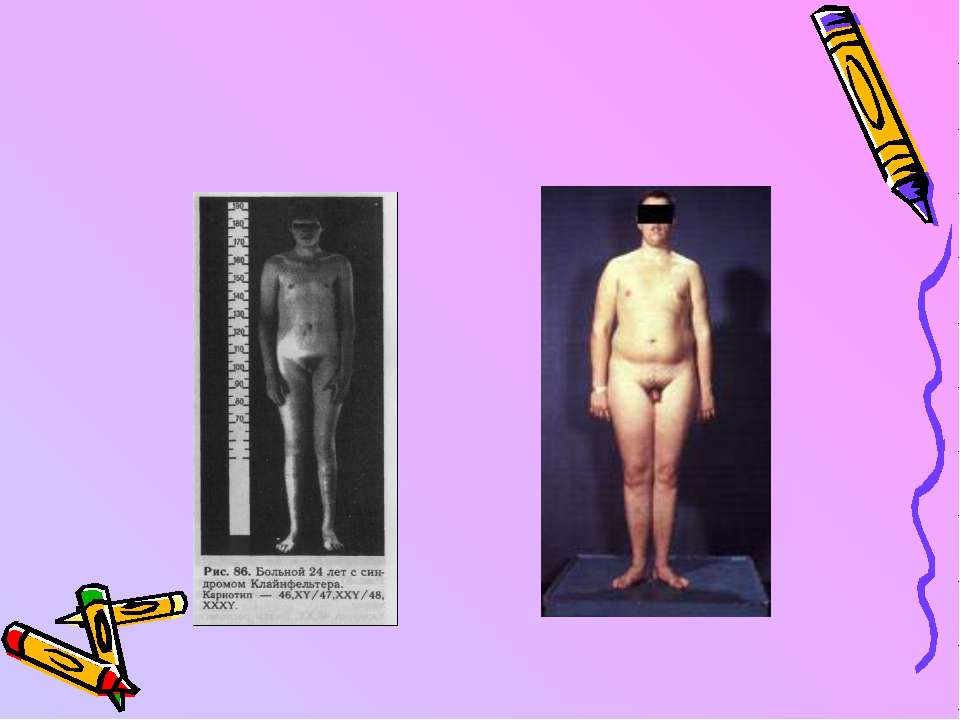

Синдром Тернера - це гіпогонадизм у жінок. Частота його - 1/1250. Каріотип - 45,ХО. Він виникає при нерозходженні Х-хромосом в процесі мейозу. Тоді одна дочірня статева клітина буде містити дві Х-хромосоми, а друга - жодної. При заплідненні їх з нормальними ооцитами утворяться жіночі організми з трисомією або моносомією (синдромом Тернера).

Karyotype - 45,X

С-м Шерешевського-Тернера

Хворі мають коротку шию, широку грудну клітку, короткі товсті ноги, вкорочені пальці, а також фізичні вади - стеноз аорти, незарощення міжшлуночкової перегородки, підковоподібну нирку. Обов’язково наявні такі ознаки, як недорозвиток статевих органів, відсутність грудних залоз, аменорея, інфантилізм. Характерно, що не страждає або дуже мало страждає інтелект. Тому діагноз ставлять не в ранньому дитинстві, а через багато років, коли виявляється відставання росту і статевий інфантилізм. Частина хворих з синдромом Тернера - мозаїки 46,ХХ/45,ХО.


Синдром Х-трисомії зустрічається з частотою 1/1000. Каріотип - 47,ХХХ. Характерні ознаки - інфантилізм, аменорея, депігментація ділянок шкіри і волосся, розумовий недорозвиток. Сприяють виникненню синдрому старший вік матері, сифіліс, алкоголізм. Описані сім’ї, де Х-трисомія передається по спадковості.

Молекулярні спадкові хвороби спричинюються генними мутаціями.

Добре відомий патогенез фенілкетонурії. В організмі людини амінокислота фенілаланін під впливом ферменту фенілаланінгідроксилази перетворюється в тирозин, а потім - у меланін, тироксин, адреналін. При відсутності фенілаланінгідроксилази фенілаланін не може перетворитися в тирозин, він нагромаджується в тканинах і перетворюється в фенілпіровиноградну і фенілоцтову кислоти. Сам фенілаланін і обидві кислоти дуже токсичні для головного мозку на стадії його формування. В результаті виникає розумова відсталість. Діти народжуються зовні здоровими, нормально набирають масу, а в другому півріччі у них виявляється відставання в психічному розвитку. Потім приєднуються інші симптоми - фізичний недорозвиток, затримка прорізування зубів, затримка мови, судороги, парези. У таких дітей зменшується утворення меланіну, адреналіну і тироксину. Тому в них світле волосся, голубі очі і артеріальна гіпотонія. Хвороба передається за аутосомно-рецесивним типом. Патологічний ген локалізований у 12 хромосомі. Частота хвороби - 1/20000.

Альбінізм розвивається внаслідок відсутності пігменту меланіну. Він може бути загальним і місцевим (наприклад, очним). Хвороба зумовлена дефіцитом ферменту тирозинази, яка перетворює тирозин у меланін. Передається за аутосомно-рецесивним типом, частота її - 1/20000.

Алкаптонурія - також результат блокування обміну тирозину на рівні гомогентизинової кислоти. Через брак відповідної оксидази ця речовина не піддається дальшим перетворенням, нагромаджується в організмі і виділяється з сечею. В лужному середовищі сеча темніє. Відкладання гомогентизинової кислоти у сполучній тканині дає темне забарвлення - охроноз.

Із генних захворювань, пов’язаних з порушенням вуглеводного обміну, добре вивчена галактоземія. Вона розвивається тому, що галактоза не може перетворитися в глюкозу через відсутність відповідного ферменту. В організмі нагромаджується доблоковий метаболіт - галактозо-1-фосфат. Він відкладається в різних органах - кришталику, печінці, мозку, нирках і порушує їх функцію. Симптоми хвороби проявляються дуже рано. через кілька днів від початку годування молоком (втрата апетиту, діарея, жовтяниця, збільшення селезінки, асцит). Дуже характерні катаракти, що розвиваються на третьому тижні життя. Затримується ріст і розумовий розвиток. Можливий смертельний кінець.

Головних типів три - аутосомно-домінантний, аутосомно-рецесивний і Х-зчеплений (рецесивний або домінантний).

Аутосомно-домінантним називають такий тип спадкування, коли хвороба передається з покоління в покоління - від батьків до дітей, від дітей до внуків і т.д. Труднощів для діагностики такі хвороби, як правило, не викликають. За аутосомно-домінантним типом спадкуються фізичні аномалії - короткопалість, ліворукість, близько- і далекозорість, а також хвороби - вроджена катаракта, отосклероз, поліпоз товстої кишки, вроджена гемералопія, хорея Гентінгтона.

Аутосомно-рецесивним називають такий тип спадкування, коли хвороба передається не прямо, а через одне чи декілька поколінь (з пропусками). Хвороба проявляється тільки у гомозигот, а поява їх найчастіше пов’язана з близькородинними шлюбами. За таким типом передаються вроджена глухонімота, шизофренія, фенілкетонурія, альбінізм, алкаптонурія, кретинізм, гіпофізарна карликовість.

Коли патологічний ген знаходиться в Х-хромосомі, то кажуть, що спадкування Х-зчеплене. Як правило, ген проявляє себе рецесивно. Хворіють лише чоловіки з даної родини, а жінки виконують роль кондукторів (провідників) патологічного гена. Так спадкуються гемофілії А і В, дальтонізм, юнацька глаукома, агамаглобулінемія, атрофія зорових нервів, деякі міопатії. Дуже рідко патологічний ген, що знаходиться в Х-хромосомі, проявляється домінантно (вітамін D-резистентний рахіт, фолікулярний гіперкератоз).

Спадкові хвороби вивчають і діагностують за допомогою спеціально розроблених методів

Цитогенетичний метод полягає у дослідженні особливостей хромосомного набору і будови окремих хромосом. Зручним об’єктом для цього служать лейкоцити і клітини епітелію щоки. Крім кількості і структури хромосом, досліджують статевий хроматин. Якщо в клітині є дві Х-хромосоми, то одна з них спіралізується і цей утвір стає помітним під мікроскопом. Його називають статевим хроматином, або тільцем Бара. В нормі одне тільце Бара можна побачити в жінок. При синдромі Тернера тільце відсутнє, при Х-трисомії їх помітно два, при синдромі Кляйнфельтера вони з’являються у чоловіків. Ще один об’єкт цитогенетичного дослідження - ядра нейтрофілів. У жінок частина з них має вирости, названі барабанними паличками. В патології кількість їх може зростати, що також відображає збільшення кількості Х-хромосом.

Генеалогічний метод полягає у вивченні спадкових властивостей людини за її родоводом. Метод дає цінну інформацію в тих випадках, коли відомі прямі і непрямі родичі пробанда - носія патологічної спадкової ознаки. Метод розроблений Ф.Гальтоном і вперше застосований ним для вивчення закономірностей спадкування розумових здібностей людини.
, якому вист...")
Складання родоводу починається з обстеження хворого (пробанда), якому виставляють певний діагноз. Далі користуються розповідями родичів, оглядом членів сім’ї, історіями хвороби, архівними даними. За допомогою цього методу можна визначити тип спадкування хвороби.

Близнюковий метод - один з головних методів генетики людини. Він дозволяє розмежувати роль спадковості і середовища у виникненні захворювань. Суть його полягає в тому, що вивчають попарну захворюваність (конкордантність) у однояйцевих і двояйцевих близнят за певними нозологічними одиницями. Однояйцеві близнята відрізняються від двояйцевих тим, що у перших генотип тотожний, а в других - лише схожий (як у братів і сестер). Якщо захворювання спадкове, процент конкордантності (збігу хвороб) у однояйцевих близнят буде значно вищим, ніж у двояйцевих, тобто однаковий дефект генотипу викликатиме однакову патологію. Коли ж хвороба екзогенна, то процент конкордантності буде близьким в обох типів близнят.

Популяційно-статистичний метод полягає у вивченні частоти, з якою в даній популяції зустрічається той чи інший ген. Таким способом визначають генетичну структуру популяції. Частоту появи хвороби в родині порівнюють з частотою її в популяції.
 запобігання впливу фізичних та хімічних мутагенів, н...")
профілактичні заходи: а) запобігання впливу фізичних та хімічних мутагенів, належний гігієнічний контроль на хімічних виробництвах, всебічне обстеження нових лікарських препаратів; б) попередження небажаного дітонародження, якщо сім’я обтяжена спадковою хворобою або за допомогою амніоцентезу (дослідження амніотичної рідини) виявлений серйозний хромосомний дефект; в) роз’яснювальна робота серед молоді про небажаність близькородинних шлюбів, які стають умовою прояву рецесивних патологічних генів; г) рекомендації щодо народження дітей у більш ранньому віці, тому що з роками збільшується кількість хромосомних і генних мутацій, а значить зростає вірогідність появи спадкових хвороб.

В минулому вважалося, що спадкова ознака не піддається корекції, а тому лікувати спадкові хвороби безперспективно. Помилковість цих тверджень стала очевидною тоді, коли були з’ясовані глибокі біохімічні механізми спадкової патології. В принципі формування спадкової ознаки можна відкоригувати, впливаючи на всіх рівнях дії гена, але теоретично найбільш повним був би вплив на рівні генетичного матеріалу, тобто ДНК. Цим займається генна інженерія (генна терапія), яка базується на досягненнях молекулярної біології. Метод знаходиться на стадії експериментальних розробок, але має багатообіцяюче майбутнє. Для лікування спадкових хвороб давно використовується замісна білкова або ферментна терапія. Класичний приклад - лікування гемофілії А введенням антигемофільного глобуліну (фактора VІІІ).

Видалення метаболіта перед блокованим етапом - також давно апробований метод. Якщо якась харчова речовина, яка є субстратом дефектного фермента, не виробляється в організмі, а надходить з їжею, то лікування полягає в обмеженні її споживання. Вперше успішну корекцію такого типу здійснено у хворих з фенілкетонурією. Суть терапії полягає в заміні продуктів харчування, багатих на фенілаланін, гідролізатами з мінімально необхідним вмістом цієї незамінної амінокислоти. Дієтотерапію починають з перших тижнів життя під контролем рівня фенілаланіну в крові. Тривалість лікування - 5-10 років. Аналогічна терапія (до 3-х років) використовується при галактоземії. Із харчування вилучають молоко (галактозу) і замінюють його безгалактозними сумішами.

В тому разі, коли внаслідок ферментативного блоку не синтезуються кінцеві метаболіти, їх компенсують введенням ззовні. При кретинизмі вводять тироксин, при порушенні утворення урацилу, необхідного для синтезу ДНК, до їжі додають уридин і цим запобігають проявам хвороби.

Більшість спадкових хвороб лікують шляхом усунення побічних симптомів. Цей підхід не вимагає точного знання генетичних і патофізіологічних механізмів їх розвитку. Наприклад, ми майже нічого не знаємо про молекулярні основи полідикталії, заячої губи або розщелини піднебіння, але це не заважає успішно лікувати їх хірургічним шляхом. Дуже мало відомо про біохімію психічних захворювань, хоча емпіричним шляхом вдається підібрати препарати для медикаментозного лікування.





Схожі презентації











Категорії