Презентація на тему:
Спадкові хвороби людини.
Завантажити презентацію






 Фенілкетонурія Описана в...")

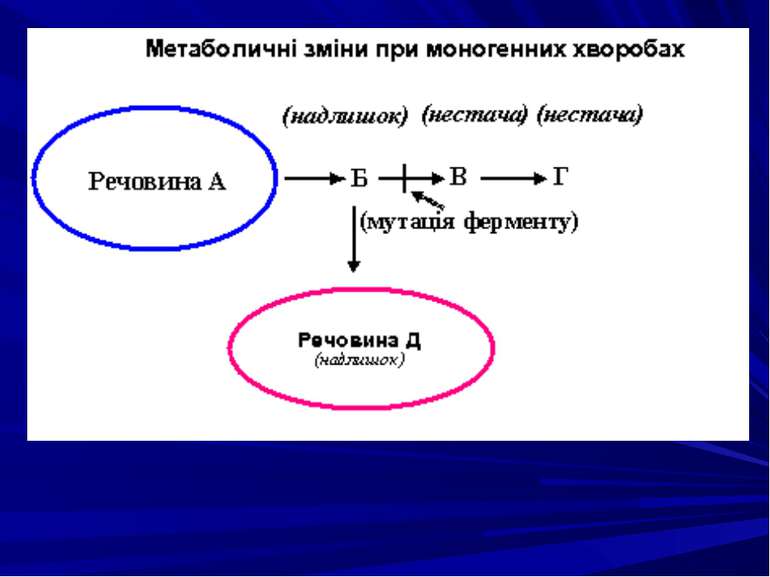
: Дефект тирозин–амінотрансферази або оксидази n–гідро...")
 Частота в популяції від 1:5000 до 1:25 000 Гетерогенність Тир...")
 Мутації трьох різних ферментів (декарбоксилази, трансаци...")
 Дефект оксидази, яка каталізує перетворення гемогентензино...")

 –– порушення обміну галактози, яка надходить з їжею та у...")
 проявляється жовтяниця новонароджених, блювання, пронос...")
 Форми: Печінкова М язова Генералізована")

 проявляються в порушенні розвитку: карликовість, особли...")




 Починається в 4-6 місяців; Порушення зору: зниження ...")










 и печінки (2) Рис. 1 Рис. 2")
 –– група спадкових хвороб, при яких уражаються п...")

")




, синдром Клайнфельтера (1942), синдро...")

, трисомія D, синдром трисомії 13 – 15 хромосоми, ...")
 прояви: мікрокранія, аномалії мозку, анофтальм, заяча г...")


 трисомія 18, трисомія Е (по 17…18 хромосомам...")
 трисомія 21. Каріотип 47 хромосом. Частота 1: 700…...")



, делеція короткого...")

, каріотип...")







Спадкові хвороби людини.
Завантажити презентаціюПрезентація по слайдам:

Спадкові хвороби людини.

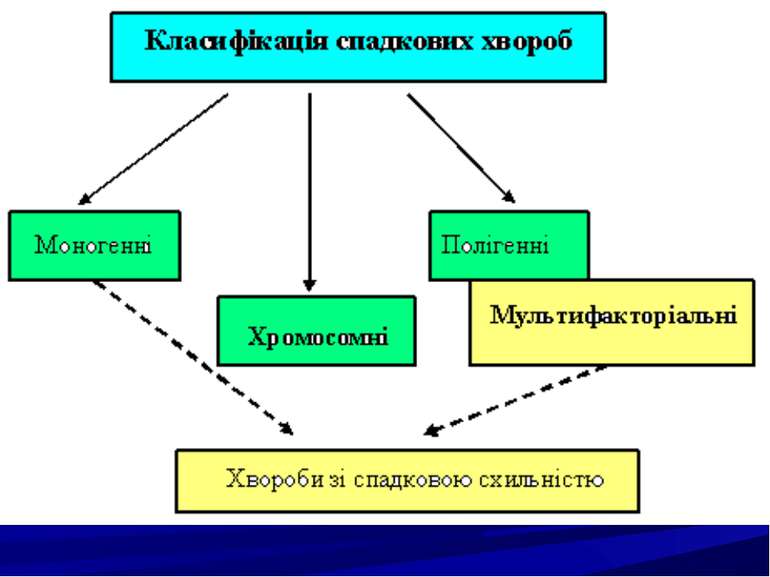
Спадкові хвороби людини, їх класифікація. Молекулярні спадкові хвороби. Хромосомні хвороби. Мультифакторіальні хвороби. Хвороби із спадковою схильністю. Принципи діагностики спадкової патології.
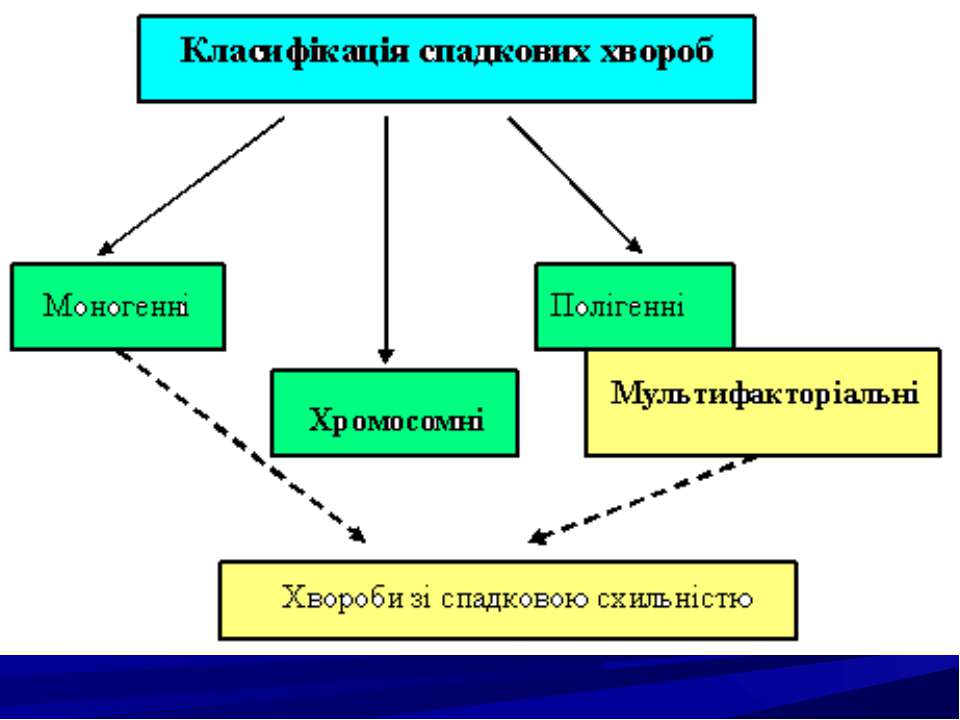
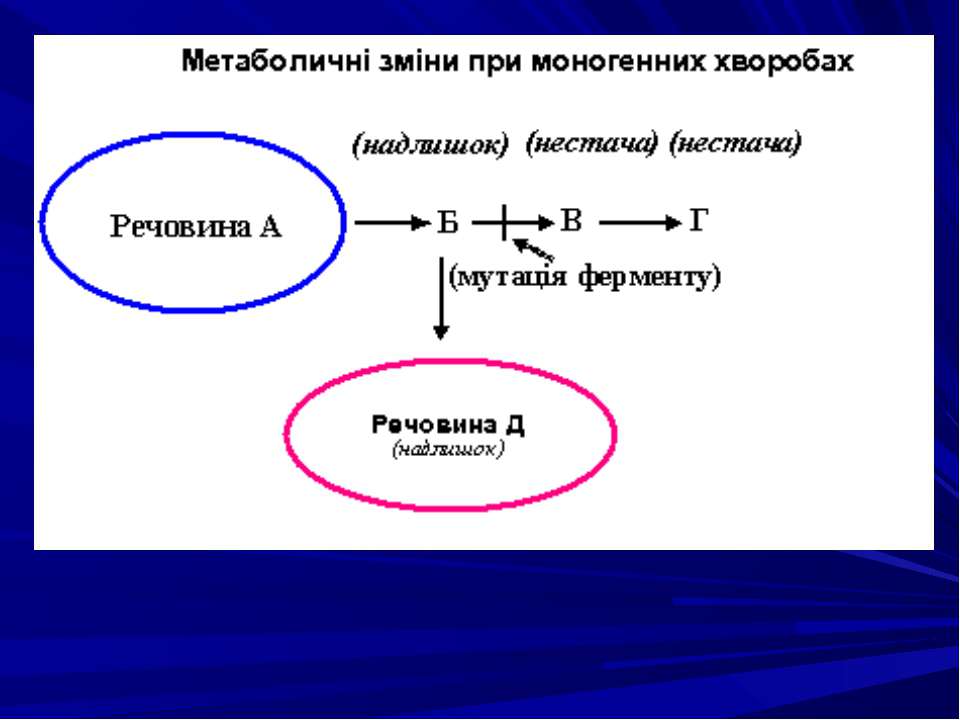

Моногенні хвороби поділяються на групи залежно від характеру порушень: ензимопатії (ферментації); дефекти структурних білків і транспортних; порушення циркулюючих білків крові; генні хвороби з невідомим первинним біохімічним дефектом.

Моногенні хвороби – порушення обміну амінокислот фенілкетонурія Тирозиноз Альбінізм Алькаптонурія Лейцинози Гістидинемія Гомоцистинурія

Фенілкетонурія А-Р 1 : 30 000 1: 6 000
 Фенілкетонурія Описана в...")
МХ порушення амінокислотного обміну (аміноцидопатії) Фенілкетонурія Описана в 1934р., висока частота захворюваності (1:10000...40000) Гетерогенність Дієторезистентні форми (генокопії) Варіабельна експресивність Дефект фенілаланін – 4 – гідроксилази

Клінічні прояви неврологічного характеру: підвищена збудливість, гіперрефлексія та підвищений тонус м’язів, тремор, судомні епілептоїдні напади, пізніше розвивається мікроцефалія, розумова відсталість, зменшена пігментація шкірних покривів, волосся, райдужної оболонки очей.
: Дефект тирозин–амінотрансферази або оксидази n–гідро...")
Тирозиноз (тирозинемія): Дефект тирозин–амінотрансферази або оксидази n–гідроксипіровиноградної кислоти Накопичення в крові та виділення з сечею тирозина. Порушення гомеостазу. В гострій формі захворювання характеризується затримкою розвитку малюка, появою гепатоспленомегалії, геморагії, змінами в нирках. Без лікування діти гинуть в грудному віці від печінкової або дихальної недостатності. Для хронічного перебігу хвороби притаманні цироз печінки, рахітоподібні зміни кісток, ураження канальцевої системи нирок.
 Частота в популяції від 1:5000 до 1:25 000 Гетерогенність Тир...")
Альбінізм (А-Р) Частота в популяції від 1:5000 до 1:25 000 Гетерогенність Тирозиназа, каталізує синтез меланіну з тирозину через 3, 4 – дигідро–ксифенілаланін. Відсутність меланіну в клітинах шкіри, волосся та райдужної оболонки очей та підвищення чутливості до УФ – випромінення.
 Мутації трьох різних ферментів (декарбоксилази, трансаци...")
Лейциноз (генокопії) Мутації трьох різних ферментів (декарбоксилази, трансацилази, флавінового ферменту), порушується окислювальне декарбоксилювання 3–х кетокислот, в які перетворюється лейцин, ізолейцин і валін. Клінічно розрізняють декілька форм цієї хвороби –– класичну, проміжну, м’яку, тіамінзалежну. Основні симптоми пов’язані з ураженням нервової системи: судоми, порушення дихання, в сечі надлишок кетокислот надає їй запаху кленового сиропу.
 Дефект оксидази, яка каталізує перетворення гемогентензино...")
Алкаптонурія (А-Р) Дефект оксидази, яка каталізує перетворення гемогентензинової кислоти в малеїнацетооцтвову. Клінічні прояви: починаються після 40 років, патологія суглобів, кінцівок та хребта, інших збагачених сполучною тканиною частин тіла. Відкладання гомогентинзи-нової кислоти в сполучній тканині (пігментація кольору охри). Велика кількість кислоти виводиться з сечею (потемніння на повітрі).

Моногенні хвороби – порушення обміну вуглеводів Галактоземія, Фруктоземія, Глікогенози, Мукополісахаридози.
 –– порушення обміну галактози, яка надходить з їжею та у...")
Галактоземія (А – Р) –– порушення обміну галактози, яка надходить з їжею та утворюється при гідролізі лактози. у гомозигот активність ферменту 3–12 % від норми, у гетерозигот –– 50 %. Частота 1:35…150 тис. народжень. характеризується гетерогенністю. Н., з частотою 1:100…200 тис. зустрічається галактоземія з м’якою клінічною картиною.
 проявляється жовтяниця новонароджених, блювання, пронос...")
Фенотипово (клінічно) проявляється жовтяниця новонароджених, блювання, пронос, розвиток розумової відсталості, враження печінки, дистрофія. При ранній діагностиці дитині призначають спеціальну дієту –– виключення молока матері та інших продуктів, які містять лактозу або галактозу. Розвиток нормалізується.
 Форми: Печінкова М язова Генералізована")
Глікогенози А-Р (1 : 40 000) Форми: Печінкова М язова Генералізована

Мукополісахаридози – хвороби накопичення – спадкові хвороби – порушення метаболізму глікозамінгліканів (ГАГ), накопичення ГАГ внаслідок мутацій ферментів лізосом (гідролаз). Генетична гетерогенність визначається мутаціями різних генів, які кодують різні ферменти. Тип успадкування А – Р, Х – Р.
 проявляються в порушенні розвитку: карликовість, особли...")
Фенотипово (клінічно) проявляються в порушенні розвитку: карликовість, особливі риси обличчя, малорухомість суглобів, зменшення мозку. Рання смертність – 12–20 років. З сечею виділяється багато мукополісахаридів. Найчастіше зустрічається синдром Гурлера (гаргоілізм), синдром Хантера (мукополісахаридоз, тип ІІ).

Гаргоїлізм хвороба Гурлера

Спадкові дефекти обміну ліпідів –– сфинголіпідози –– порушення розщеплення ліпідів та обміну ліпідів плазми крові. Тип успадкування А – Р, Х – Р. Частота різних форм від ≈ 1:4000 новонароджених до 1: 300 000, частота в різних популяціях може значно відрізнятися.

Накопичення ліпідів у внутрішніх органах. Початкові симптоми: відказ від їжі, блювання, в подальшому зниження ваги, гіпертрофія внутрішніх оргвнів, затримка психічного розвитку. Напівлетальна мутація (смерть до 5 років) Хвороба Німанна-Піка

Ураження внутрішніх органів при генералізованої формі хвороби
 Починається в 4-6 місяців; Порушення зору: зниження ...")
Хвороба Тея- Сакса (А-Р) Починається в 4-6 місяців; Порушення зору: зниження зору, симптом “вишневої кісточки”,атрофія зорових нервів, сліпота; Зникають орієнтувальні та захисні реакції, підвищені реакції на звукові сигнали; Смертність через 1-2 роки після початку.

Спадкові хвороби пуринів і пиримидінів. Приклад синдром Леша – Найяна. Частота 1 : 300000. Тип успадкування може бути Х – Р, А – Р. Нестача ферменту, необхідного для синтезу ДНК (гіпоксантин-фосфорибозилтрансферази). В сечі хворих накопичується сечова кислота. Фенотипові порушення: розумова відсталість, симпатичні паралічі, підвищена збудливість, порушення пуринового обміну, агресивна поведінка, нирковокам’яна хвороба (накопичення уратів).

Гемоглобінопатії Гемоглобінопатії –– група спадкових хвороб, при яких порушуються білкові ланцюги гемоглобіну (Нb), що призводить до змін їх функцій і властивостей. До таких хвороб відносяться: метгемоглобінемія, еритроцитози, серпоподібно–клітинна анемія, таласемія.

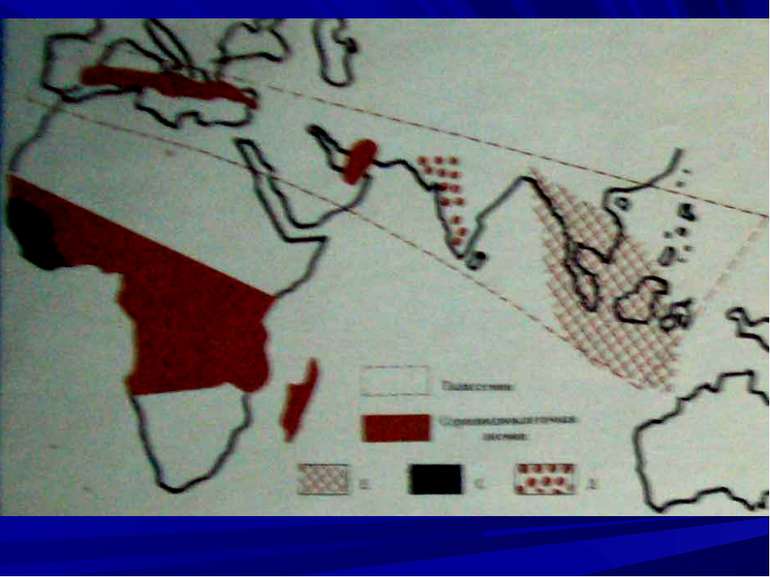
Серпоклітинна анемія З високою частотою зустрічається в регіонах розповсюдження малярії. Тип успадкування –– аутосомний, неповністю домінантний. Мутантний ген (S) викликає синтез гемоглобіну S, який змінює форму еритроцитів та слабо приєднує кисень, в наслідок чого розвивається анемія та гіпоксія. У гетерозигот –– одночасно є нормальний Нb та мутантний НbS, але вони не хворіють на малярію.

Таласемії –– хвороби, при яких зменшується вміст білку – глобіну в молекулі гемоглобіну (Hb). Тип успадкування А – Р, або внаслідок делецій. Для діагностики виду таласемій використовують молекулярно–генетичний метод, електрофорез.

Серпоклітинна анемія Таласемія

Колагенові хвороби. В основі цих хвороб генетичні дефекти біосинтезу та розпаду колагену (структурний компонент сполучної тканини). До цієї групи відноситься хвороба Елерса – Данлоса, для якої характерний генетичний поліморфізм, тип успадкування А – Д, А – Р; хвороба Марфана (А – Д тип успадкування).

Фенотипово плейотропна дія мутантних генів проявляється: гіпермобільним синдромом, збільшеною еластичністю шкіри, внутрішніми кровотечами, змінами в суглобах, блакитними склерами. Первинні дефекти –– порушення біосинтезу колагену або процесингу фібрил і колагену.

Синдром Марфана – арахнодактилія

Ахондроплазія А-Д А–Д тип успадкування, частота 1:100 000; виникає внаслідок мутації (de novo). Фенотипово проявляється порушеннями скелету (порушення утворення хрящової тканини в епіфізах трубчастих кісток, кісток черепу).

Муковісцидози (А – Д або А – Р тип успадкування, частота 1:2500 новонароджених). В основі патогенезу усіх форм –– ураження ендокринних залоз (секретуючих клітин бронхів, підшлункової залози, кишечнику, потових залоз, печінки) відбувається виділення густого секрету, запальним та склеротичним змінам в органах. Основні форми –– печінкова, легенева та кишкова. Діагностика –– спеціальні комплексні тести – визначення вмісту Na+ в секретах, визначення активності травних ферментів.
 и печінки (2) Рис. 1 Рис. 2")
Зміни структури піджлункової залози (1) и печінки (2) Рис. 1 Рис. 2
 –– група спадкових хвороб, при яких уражаються п...")
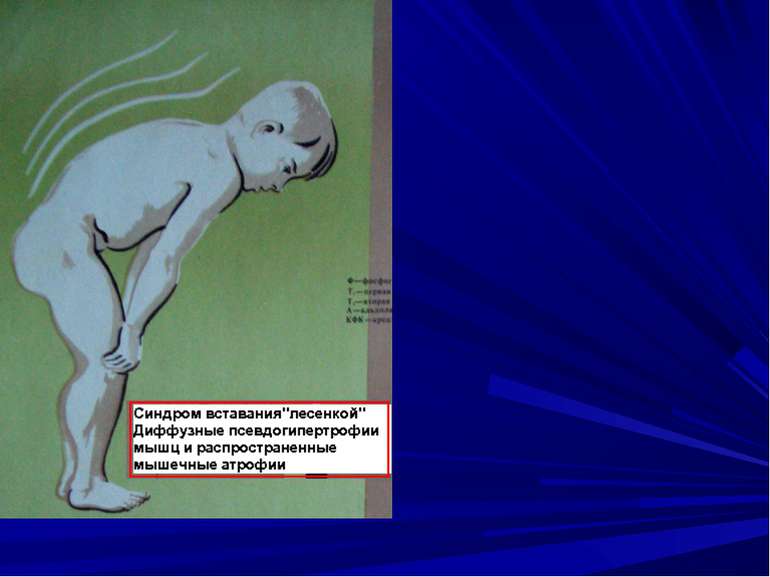
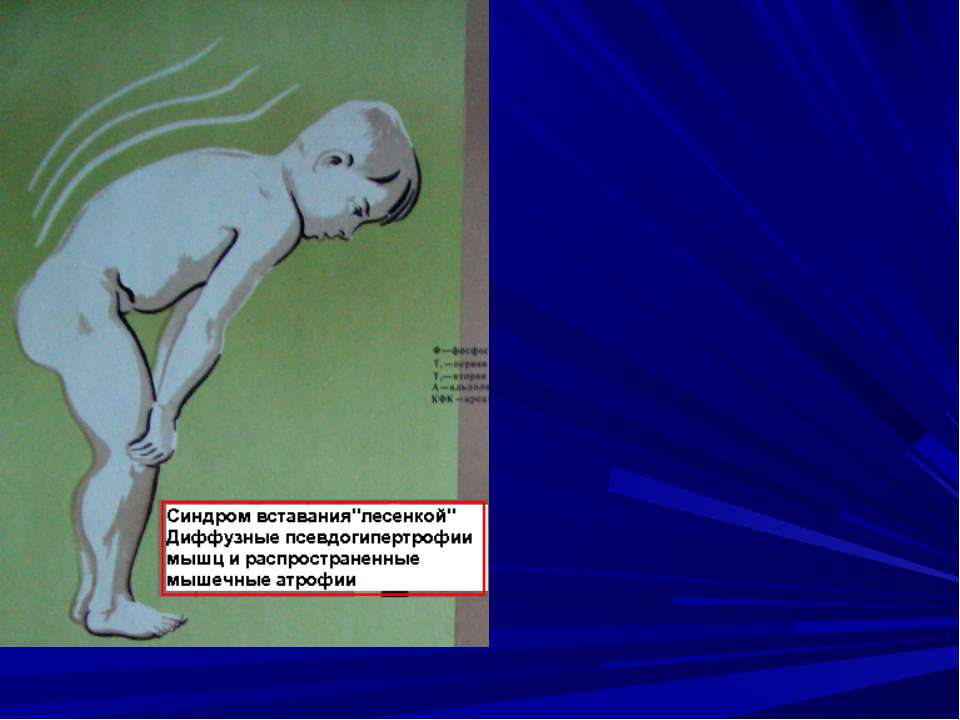
Міопатії (м’язові дистрофії) –– група спадкових хвороб, при яких уражаються посмуговані та гладенькі м’язи. Тип успадкування може бути Х – Р, А – Д, А – Р. Для міопатій характерне ураження м’язів, яке з віком прогресує, клінічний поліморфізм.

Ювінільна форма міопатії Атрофия м’язових волокон та кордіосклероз Межреберні м’язи – некроз и атрофія
")
Мультифакторіальні хвороби (МХ)

Характеризуються безперервним рядом фенотипових змін від субклінічних до чітко виражених клінічних симптомів. Велике значення в етіології, розвитку, прояву МХ має вплив середовищних факторів, особливе значення при цьому відіграють випадкові фактори, які значно стимулюють експресивність ознак при МХ та стимулюють ранню маніфестацію та швидкий прогрес патології.

При клініко–генеалогічному аналізі МХ враховують: частоту захворювання у родичів, сибсів, яка може бути значно більшою ніж загально-популяційна; залежність частоти захворювання від статі, а також від лінії, якою передається (частіше за материнською).

Хвороби із спадковою схильністю можуть бути моногенними. Наприклад, існує спадкова патологія –– нездатність переносити лактозу. Дані гени широко розповсюджені серед населення Азії (95 – 100%) та в деяких інших популяціях; підвищена чутливість до сульфаніламідних препаратів, яка призводить до гемолізу еритроцитів, підвищенню температури.

До мультифакторіальних хвороб відносяться такі хвороби, як псоріаз, цукровий діабет, шизофренія, гіпертонічна хвороба. Часто схильність до ряду хвороб спостерігається у людей з певними генотипами за полігенними системами груп крові, HLA (хела) та іншими.
, синдром Клайнфельтера (1942), синдро...")
Хромосомні хвороби хвороба Дауна (1866), синдром Клайнфельтера (1942), синдром Шерешевського – Тернера (1925, 1938). Встановлення зв’язку міх хворобами і змінами кількості хромосом було доведено тільки у 1959 р. 500 хромосомних хвороб –– порушень кількості та структури хромосом.

Схема нерозходження однієї пари хромосом у 1-му мейотичному поділі. Внаслідок злиття аномальної гамети з нормальною гаметою утворюються зиготи з трисомією (1) або моносомією (2). А – перший і другий мейотичний поділ; Б - зиготи: 1 – з трисомією; 2 - з моносомією. 2n+1 2n-1
, трисомія D, синдром трисомії 13 – 15 хромосоми, ...")
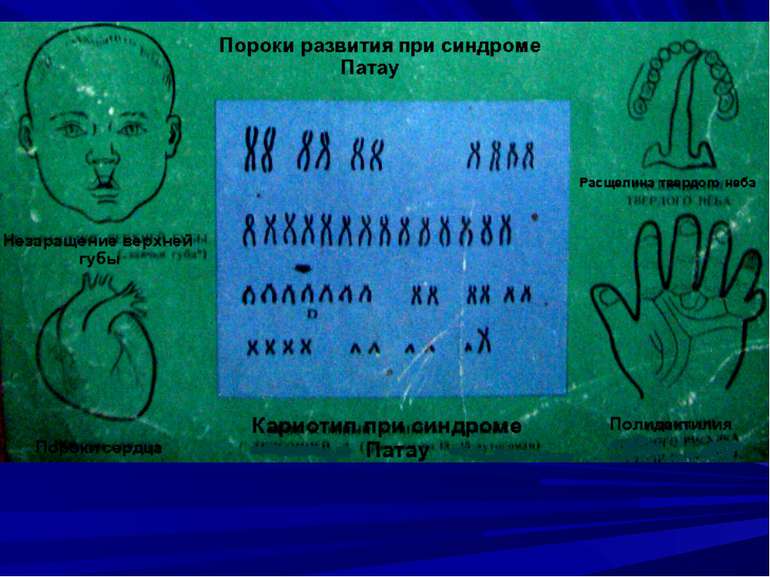
Синдром Патау (Patau, 1960), трисомія D, синдром трисомії 13 – 15 хромосоми, частіше 13 хромосоми. Частота 1: 5000…7000 народжень.

 прояви: мікрокранія, аномалії мозку, анофтальм, заяча г...")
Фенотипові (клінічні) прояви: мікрокранія, аномалії мозку, анофтальм, заяча губа, вовча паща, полідактилія, гемангіоми (аномалії внутрішніх органів) багаточисельні, вроджені вади серця. Зміни дерматогліфіки (тупий кут аtd), поперечна долонна складка. S–подібна фабулярна дуга в ділянці великого пальця стопи. Висока дитяча смертність (90% протягом року).

Синдром Едварса Трисомія 18

Кариотип при синдромі Едварса
 трисомія 18, трисомія Е (по 17…18 хромосомам...")
Синдром Едвардса (Edwards, 1960) трисомія 18, трисомія Е (по 17…18 хромосомам). Частота ≈ 1: 7000 народжень. Фенотипово проявляється комплексом варіабельних вад розвитку. Недостатній фізичний та психічний розвиток. М’язові атрофії, глибоке розташування та дисплазія вушних раковин. Порушення розвитку очей: мікрофтальм, аномалії райдужної оболонки, епікант. Аномалії скелету, крилоподібна шкіра шиї, щитоподібна форма грудної клітки, дисплазія грудини, таза, аномалії ребер, сколіоз, множинні вади розвитку внутрішніх органів (серця, легень, нирок), гіпоспадія. Діти гинуть протягом 1–го –– 2–х років.
 трисомія 21. Каріотип 47 хромосом. Частота 1: 700…...")
Синдром Дауна (Dawn, 1866) трисомія 21. Каріотип 47 хромосом. Частота 1: 700…800 новонароджених, частота зростає з віком матері (після 36 – 40 років становить 1: 50). Результат трисомії 21 хромосоми, транслокації або мозаїцизму.

Синдром Дауна

Каріотип 47,21+

Синдром Дауна Вади розвитку
, делеція короткого...")
Синдром “котячого крику” (синдром „cri – du – chat”; 1963), делеція короткого плеча 5 хромосоми (5р–). Частота серед новонароджених 1: 40000…50 000. Характеризується поліморфізмом в залежності від розміру делеції. Фенотипово спостерігається недостатній фізичний та розумовий розвиток, аномалій внутрішніх органів, мікроцефалія. Під час народження характерний котячий крик.

Гоносомії
, каріотип...")
Синдром Шерешевського – Терненра (Шерешевський, 1925; Turner, 1938), каріотип 45, ХО (моносомія Х). Частота 0,7 : 1000 дівчаток.

Діагностика Тільця Барра Барабанні палички Зміни дерматогліфіки

Синдром трипло-Х Х – полісомія зустрічається у жінок. Каріотип 47, ХХХ або випадки мозаїцизму на 46, ХХ/ 47, ХХХ. Частота 1: 1000 народжень.

Синдром Клайнфельтера (Altmann, 1895, каріотип 47, ХХУ; 48, ХХХУ; 49, ХХХХУ. Kleinfelter і співавтори,1942). Частота 1,5 : 1000 народжених хлопців, інші варіанти –– рідко

Каріотип

Діагностика Барабанні палички Тельца Барра – Х-хроматин

Фенотиповий прояв. Юнаки високого росту, з непропорційно довгими кінцівками, генікомастією, із зниженням лібідо. Порушується статевий розвиток, гіпоплазія яєчок (гістологічно – звуження або повна облітерація сім’яних канальців). Безплідність. Психічні порушення, при збільшенні кількості Х–хромосом –– олігофренія різного ступеня. Зміни дерматогліфіки – дистальне зміщення аксіального трирадіуса і збільшення кута atd. На пальцях переважають дуги.

Діагностичне значення статевого хроматину
Схожі презентації











Категорії