Презентація на тему:
Біохімія м язів
Завантажити презентацію









 У диску А містяться товсті (міозинові) нитки т...")




 У 1985 р виявили, що рослинний алкалоїд ріано...")





 АТФ - утворюється при легкій і помірн...")












Біохімія м язів
Завантажити презентаціюПрезентація по слайдам:

Біохімія м’язової тканини

Функція – забезпечення рухливості шляхом скорочення і наступного розслаблення зі здійсненням роботи, пов’язаної з перетворенням хімічної енергії у механічну.

Класифікація м’язової тканини 3 типи м’язової тканини – скелетні м’язи; серцевий м’яз; гладенькі м’язи. Інший поділ – гладенькі та поперечносмугасті м’язи. До поперечносмугастих належать скелетні м’язи, м’язи язика і верхньої третини стравоходу, зовнішні м’язи очного яблука і ряд інших. Міокард за морфологічними ознаками належить до цієї ж групи; за іншими ознаками займає проміжне положення між гладенькими та поперечносмугастими. Гладенькі м’язи – це м’язи внутрішніх органів, які оточують пустотілі або трубчасті органи, які змінюються у об’ємі (є у шлунку, кишках, матці, сечовому міхурі, кровоносних судинах).

Поперечносмугасті м’язи. М’язове волокно. Міофібрили Поперечносмугасті м’язи побудовані з м’язових волокон. М’язове волокно – це багатоядерна клітина гігантських розмірів: діаметр 10-100 мкм, довжина часто відповідає довжині м’язу. Вона покрита еластичною оболонкою – сарколемою. У саркоплазмі вздовж волокна у формі пучків розташовані нитковидні утворення – міофібрили товщиною менш 1 мкм, мітохондрії, рибосоми, саркоплазматична сітка, вакуолі, глибки глікогену, включення ліпідів. Міофібрили побудовані з білкових філаментів (ниток) 2-х типів – товстих і тонких - на міофібрилу припадає біля 2 500 філаментів, причому на 1 товсту нитку припадає 2 тонкі

Поперечносмугасті м’язи. Товсті філаменти. Міозин. Товсті філаменти складаються з паралельно розташованих молекул білка міозину (М) (1 філамент – 400 молекул). Кожна молекула має 2 важкі та 4 легкі п/п ланцюги. Важкі ланцюги переважно мають α-структуру і закручені один навколо іншого („хвіст” молекули); її „голівка” - це кінець кожного з важких ланцюгів разом із 2-ма легкими. Молекула М може згинатися так, що голівка і частина хвоста повертаються, як на шарнірі. Довжина молекули М – 150 нм; товщина – 2 нм; довжина товстих міозинових філаментів – 1,5 мкм; діаметр – 10-14 нм. М має властивості АТФази, активний центр якої локалізований у голівках молекули і містить у зв’язаному стані мол-лу АТФ. Половина з мол-л міозину звернені голівками до одного кінця філамента, друга половина – до іншого, причому по довжині філамента молекули зсунуті одна відносно одної.

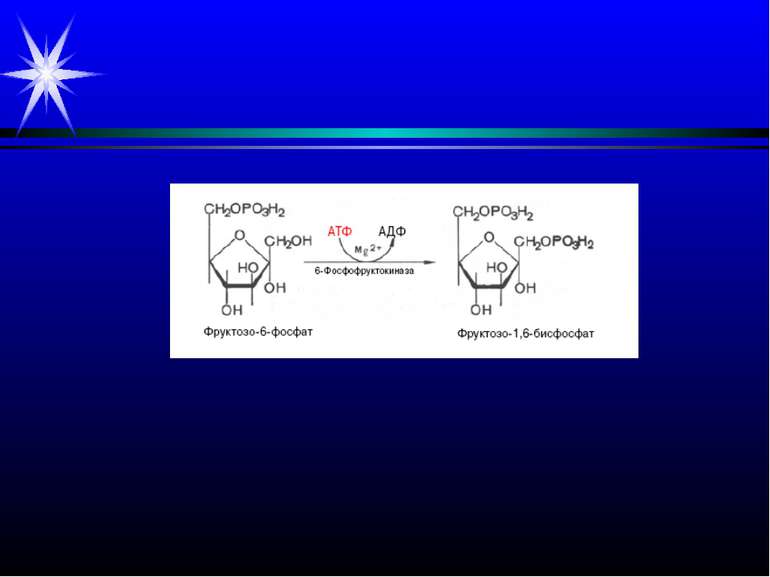
Поперечносмугасті м’язи. Тонкі філаменти. Актин. Тонкі філаменти у складі мають білки актин (А), тропоміозин (ТМ) та тропонін (ТН). Є дві форми А: глобулярний G-актин (G-А) і фібриллярний F-актин (F-А). Молекули G-А нековалентно з’єднуються і утворюють F-А. Два ланцюги F-А закручені один навколо іншого. Кожна молекула G-А має центр зв’язування міозину, який у стані спокою заблокований. У повздовжньому жолобку спіралі F-А розміщена паличкоподібна молекула білка ТМ: на 1 молекулу ТМ довжиною 41 нм припадає 7 пар G-А. Сюди ж приєднується 1 молекула глобулярного білка ТН, що складається з 3-х субодиниць (С, І, Т). Субодиниця С (кальційзв’язуюча) має спорідненість до іонів кальцію; субодиниця І (інгібуюча) – може інгібувати АТФазну активність; субодиниця Т (тропоміозинзв’язуюча) – забезпечує зв’язок із ТМ. Утворені структури (F-А + ТМ + ТН) об’єднуються між собою в тонкі філаменти довжиною 1 мкм. ТН та ТМ є регуляторними білками, що запускають і виключають утворення поперечних містків між актином і міозином.

Поперечносмугасті м’язи. Саркомер При дослідженні волокон скелетних м’язів у поляризаційному або фазово-контрастному мікроскопах виявляється поперечна смугастість волокна, яка пояснюється оптичною неоднорідністю білків, локалізованих у всіх міофібрилах на одному рівні. Елементом поперечносмугастої міофібрили, який повторюється, є саркомер Кожна міофібрила містить кілька сотен саркомерів, довжина кожного з яких – 2,5-3,0 мкм.

Будова саркомеру Саркомер - це ділянка міофібрили, межами якої є вузькі Z-лінії. Всередині саркомера міститься ділянка довжиною 1,5-1,6 мкм, яка є темною у фазово-контрастному мікроскопі та в поляризованому світлі виявляє сильне подвійне променезаломлення – диск А (анізотропний диск). В центрі диску А міститься лінія М, яку спостерігають лише в електронному мікроскопі. Середню частину диску А займає ділянка Н з більш слабким подвійним променезаломленням. Існують також диски І (ізотропні диски) з дуже слабким подвійним променезаломленням, у фазово-контрастному мікроскопі вони є більш світлими, ніж диски А; їх довжина біля 1 мкм, і кожен з них розділений на 2 рівні половини Z-смугами
 У диску А містяться товсті (міозинові) нитки т...")
Будова саркомеру (продовження) У диску А містяться товсті (міозинові) нитки та тонкі (актинові) нитки. Тонкі нитки починаються у межах кожного саркомера у Z-лінії, протягуються через диск І , проникають у диск А і перериваються у ділянці Н. Тобто ділянка Н не містить тонких ниток, а Диск І – товстих. При скороченні м’язу диски І в ній майже щезають, а ділянка перекриття товстих і тонких ниток – збільшується; при цьому саркомер вкорочується до 1,7-1,8 мкм ( від 2,5-3,0 мкм).

Хімічний склад поперечносмугастого м’язу: білки й ліпіди Білки саркоплазми – розчиняються у воді або сольових розчинах. Міогенна, глобулінова, альбумінова та міоглобінові фракції, які не є однорідними. Міогенна фракція включає ряд ферментів гліколізу та ін білки. Серед інших білків саркоплазми є білки-ферменти мітохондрій, які відповідають за клітинне дихання, ферменти азотистого та ліпідного обміну. Міоальбумін саркоплазми за хім.. властивостями нагадує альбумін плазми крові. Міоглобін м’язів – хромопротеїн, який з’єднується із киснем та забезпечує процес дихання м’язів – пояснює червоний колір м’язової тканини. Цей білок має спорідненість до кисню, у 5 разів вищу, ніж гемоглобін. Міофібрилярні білки – міозин ( 56-60%), актин (20-25%) і регуляторні білки тропоміозин ( 10-15%), тропонін (4-6%). Білки строми – колаген, нейрокератин, еластин. Вони входять до складу сполучнотканинних елементів стінок судин, нервів та сарколеми. Ліпіди - нейтральні жири, які входять у простір між структурами м’язових волокон і є резервним жиром; холестерол, фосфоліпіди – входять до складу клітинних мембран, їх вміст зростає під час тренування.

Хімічний склад поперечносмугастого м’язу. Екстрактивні азотисті небілкові речовини Аденінові нуклеотиди – АТФ, АДФ, АМФ; Нуклеотиди неаденінового ряду (ТТФ, УТФ, ЦТФ, інозинмонофосфат); Креатинфосфат, креатин, креатинін, карнозін, ансерин, вільні амінокислоти. На долю креатину та креатинфосфату припадає до 60% небілкового азоту – вони мають відношення безпосередньо до скорочення м’язів. Креатинін в основному синтезується у печінці за участю аргініну, гліцину та метіоніну; з током крові він надходить у м’язи, де фосфорилюється з утворенням креатинфосфату. Карнозин і ансерин (метильоване похідне карнозину) – імідазольні дипептиди, які підвищують ефективність роботи іонних насосів м’язової тканини, сприяють збільшенню амплітуди м’язового скорочення і проявляють антиоксидну дію. Вільні амінокислоти – це переважно глутамінова кислота та глутамін. В невеликій кількості – як проміжні або кінцеві продукти азотистого обміну – зустрічаються сечовина, сечова кислота, аденін, гуанін, ксантин та гіпоксантин.

Хімічний склад поперечносмугастого м’язу. Екстрактивні безазотисті речовини Головним чином глікоген ( 0,4-0,8%); під впливом тренування зростає до 1,5-3 %. Втомлені м’язи мають менше глікогену. Під час роботи глікоген м’язів розпадається на глюкозу, тріозофосфати та інші проміжні продукти гліколізу, у тому числі і на молочну кислоту. Слідові кількості вільної глюкози, гексозофосфатів. Мінеральні речовини –переважають катіони: натрій, калій, кальцій, магній; є мідь, марганець, цинк; і аніони – фосфати та сульфати. Їх роль – підтримка сталості рН, осмотичної рівноваги. Зниження концентрації солей веде до зменшення їх збудливості.

Скорочення м’язового волокна Ініціюється потенціалом дії, який поширюється від нейром’язового синапсу в обох напрямках вздовж м’язового волокна. Він за участю певних механізмів спричиняє зміну проникності мембран саркоплазматичного ретикулуму для іонів Са++ , які виходять у саркоплазму. У стані спокою вміст Са у саркоплазмі – 10-7 моль/л; після впливу ПД він зростає до 10-5 моль/л.
 У 1985 р виявили, що рослинний алкалоїд ріано...")
Ріанодиновий рецептор (справка) У 1985 р виявили, що рослинний алкалоїд ріанодин із кори Ryania speciosa у концентрації 10-9 – 10-8 ефективно блокує Са++-канали СР, міцно зв язуючись із невідомими білками ретикулуму, які пізніше – через 2 роки – виділили й назвали ріанодиновим рецептором (RYR). RYR-1 – ізоформа скелетних м язів; RYR-2 – серцевого м”язу, також присутня у деяких нервових клітинах); RYR-3 – у мозку, серцевому м“язі й незбудливих тканинах. RYR1 і RYR2 – 60% гомології Скорочення скелетних і серцевих м”язів активується, коли Ca++ звільнюється із СР у цитозоль через ріанодинові рецептори

RYR і стимуляція скорочення скелетних м язів При стимуляції м”язових волокон в ділянках нервово-м язового синапсу із пресинаптичних нервових закінчень виділяється медіатор ацетилхолін (АХ), який зв язується із нікотиновими холінорецепторами, розташованими на ПМ постсинаптичної м”язової клітини в ділянці синапса, які одночасно є Na-каналами, що відкриваються при зв язуванні АХ. При цьому в саркоплазму входять іони Na (за градієнтом концентрації та градієнтом заряду) і потенціал на ПМ змінюється від –80 мВ до +40 мВ, тобто відбувається деполяризація мембрани. Деполяризація ПМ спричиняє конформаційні зміни повільних потенціалчутливих Са++-каналів ПМ (= Са++-канали L-типу, =дигідропіридинчутливі Са++-канали), які при цьому відкриваються і зумовлюють надходження у ЦП м язових клітин невеликої кількості Са++ із позаклітинного середовища. В скелетних м язах Са++-канали ПМ і ріанодинові рецептори безпосередньо контактують один із одним: приблизно із половиною молекул RYR зв язано по 4 молекули Са++-каналів ПМ. Використовуючи мутантні форми Са++-каналів ПМ, не здатних пропускати Са++ через ПМ, але здатних змінювати свою конформацію під впливом деполяризації виявили, що для активації RYR достатньо лише потенціалзалежні зміни конформації Са++-каналів ПМ. Але Са++ необхідний для активації тих RYR, які не мають безпосереднього зв язку із тетрадами Са++-каналів.

RYR і стимуляція скорочення серцевого м язу В серцевому м”язі лише невелика частина RYR зв язана із Са++-каналами ПМ (1 RYR – 2 Са++-канали) і взагалі не виявлено міцних контактів RYR із ПМ. Тому в серцевому м язі діє інший механізм активації RYR – Са++-індуковане звільнення Са++. При деполяризації ПМ кардіоміоцитів активуються потенціалчутливі Са++-канали ПМ, і в цитоплазму надходить невелика кількість Са++ із позаклітинного середовища (“тригерний Са++) – її недостатньо для забезпечення м”язового скорочення, але він активує RYR, через які із ретикулуму звільнюється основна кількість Са++, потрібного для скорочення серцевого м язу

Скорочення м’язового волокна Іони Са++ приєднуються до С-субодиниці тропоніну тонких філаментів, що викликає зміну конформації білка. Це спричиняє переміщення молекули тропоміозину по жолобку тонкого філамента, в результаті на молекулах G-актину у складі F-актину відкриваються центри зв’язування з голівками міозину товстих ниток. Міозинові голівки із зв’язаними молекулами АТФ приєднуються до найближчих молекул G-актину тонких ниток, при цьому утворюються поперечні містки (3). Взаємодія актину і міозину активує АТФазний центр міозинових голівок, і АТФ розщеплюється до АДФ і Фн, які звільнюються з каталітичного центру (4). Це супроводжується зміною конформації міозину і згинанням його молекули у ділянці шарніру (4). Внаслідок такого руху тонкий філамент, з’єднаний поперечними містками з товстим, протягується вздовж останнього (4). Надалі АТФазний центр міозинової голівки зв’язує нову молекулу АТФ, що спричиняє розрив поперечних містків і відновлення вихідної конформації молекули міозину (1). Амплітуда кожного переміщення складає біля 11 нм; частота – біля 40 разів на секунду. Всі міозинові голівки рухаються одночасно, але не синхронно – це і викликає скорочення м’язового волокна.

Скорочення м’язового волокна. Схема

Розслаблення м’язу Коли нервові імпульси на волокно не надходять, вихід Са++ з саркоплазматичного ретикулуму припиняється, і Са++ із саркоплазми переноситься зворотно за рахунок Са++-АТФази мембран саркоплазматичної сітки. При зниженні вмісту Са++ у саркоплазмі до вихідного рівня комплекс Са++-тропонін дисоціює, тропоміозин зсувається по жолобку тонкого філаменту на вихідне місце і блокує на молекулах актину центри зв’язування голівок міозину. Внаслідок розриву поперечних містків волокно розслаблюється. При недостачі АТФ містки між актином і міозином не розриваються і філаменти фіксуються у з’єднаному положенні – явище контрактури м’яза, наприклад, при трупному окоченінні після смерті.
 АТФ - утворюється при легкій і помірн...")
Джерела АТФ для м’язового скорочення 1) АТФ - утворюється при легкій і помірній роботі шляхом окисного фосфорилювання (аеробно), а при тяжкій фізичній праці – за рахунок субстратного фосфорилювання при анаеробному гліколізі (з утворенням молочної кислоти із глікогену та глюкози). 2) Креатинфосфат (утворюється з АТФ та креатину): креатин +АТФ креатинфосфат + АДФ (фермент креатинкіназа) При наявності АДФ креатинфосфат віддає йому фосфатну групу, внаслідок утворюється АТФ; 3) АТФ, який утворюється при аденілаткіназній реакції: 2АДФ АТФ +АМФ Екстреним механізмом ресинтезу АТФ є креатинкіназний шлях (до 6-10 секунд інтенсивної роботи м’язів); посилення гліколізу починається з 20-ї секунди, максимум – на 40-й. При більш тривалій роботі найважливішим стає окисне фосфорилювання. Розрізняють білі та червоні м’язові волокна. Червоні мають більший вміст міоглобіну та мітохондрій – для них характерне аеробне окислення субстратів для утворення АТФ, ці м’язи скорочуються повільніше, але довго і без ознак втоми. Білі – мають більше глікогену та гліколітичних ферментів, утворюють АТФ переважно шляхом анаеробного розпаду глікогену та глюкози, характеризуються більшою АТФазною активністю міозину; ці м’язи швидко переходять від стану спокою до максимальної активності, швидше скорочуються, але раніше втомлюються. Білі і червоні волокна зустрічаються в одному м’язі.

Цикл Корі Це поєднання процесів анаеробного гліколізу в скелетних м”язах і глюконеогенезу в печінці Під час інтенсивного м”язового навантаження у м”язах утворюється надлишок молочної кислоти, яка із кров”ю надходить до печінки, де перетворюється на піровіноградну (фермент – ЛДГ) і через стадію пірувату йде на глюконеогенез Утворена глюкоза із печінки надходить у кров, а з нею – в м”язи, де під час нового фізичного навантаження знову розщеплюється під час гліколізу до молочної кислоти

Гладенькі м’язи Не містять поперечносмугастої структури, не організовані у міофібрили. Відрізняються від інших типів м’язів співвідношенням актину і міозину: вміст міозину складає лише 1/3 його у скелетних м’язах; вміст актину може вдвічі перевищувати. Актин, як і у скелетних м’язах, пов’язаний із тропоміозином; тропоніну немає. Міозин гладеньких м’язів повільно розщеплює АТФ, тому швидкість скорочення гладеньких м’язів є значно повільнішою порівняно із скелетними м’язами ( в 100-1000 разів). Ініціюють скорочення іони кальцію. При концентрації 10-5 моль/л іони кальцію зв’язуються з білком кальмодуліном, і їх комплекс активує кіназу міозину. Остання каталізує реакцію фосфорилювання легких ланцюгів міозину, після чого відбувається взаємодія голівок міозину з актиновими нитками; в результаті міоцити скорочуються. При зниженні концентрації Са в міоцитах комплекс Са-кальмодулін-кіназа дисоціює, а від міозину відщеплюються фосфорні залишки під дією фосфатази. Скорочення гладеньких м’язів не піддається „свідомому” контролю.

RYR і стимуляція скорочення гладенького м язу В гладеньких м язах, як і у серцевому, спостерігається Са++-індуковане звільнення Са++ , але у звільненні Са++ із СР беруть участь не лише RYR, але й рецептори для І3Ф. При деполяризації ПМ клітин гладеньких м язів активуються потенціалчутливі Са++-канали, і Са++, що через них входить, активує RYR. В той же час скорочення гладеньких м язів може відбуватися і без деполяризації ПМ, через вплив гормонів і фосфоінозитидний шлях. При цьому І3Ф спричиняє вихід Са++ із СР, і вже цей Са++ може активувати RYR, спричиняючи звільнення додаткових кількостей Са++.

Серцевий м’яз За будовою подібний до поперечносмугастих м’язів: містить ядра, міофібрили, побудовані з актину і міозину, мітохондрії, саркоплазматичний ретикулум. За складом міофібрилярних білків, білків строми та саркоплазматичних білків подібний до гладеньких м’язів. Вміст АТФ – 2,6 мкмоль: нижчий ніж у скелетному м’язі (4,43 мкмоль) і вищий, ніж у гладеньких м’язах (1,38). За вмістом глікогену – теж проміжне положення. Міокард багатший на фосфогліцериди, окислення яких дає значну кількість енергії для його скорочення. У роботі серцевого м’язу характерне постійне ритмічне чергування процесів скорочення і розслаблення. АТФ утворюється за рахунок окисного фосфорилювання (аеробним шляхом). При фізичному навантаженні із скелетних м’язів через кров у міокард надходить молочна кислота, яка перетворюється на піруват завдяки ферменту ЛДГ (ізофермент ЛДГ1). Потім піруват підлягає окисному декарбоксилюванню у мітохондріях. Серцевий м’яз (як і скелетний) містить ферменти окислення кетонових тіл – ці ферменти, а також ЛДГ, запобігають закисленню крові. Синтезований при окисному фосфорилюванні у мітохондріях АТФ переноситься транслоказою через внутрішню мембрану мітохондрій. Із внутрішньою стороною зовнішньої мембрани мітохондрій зв’язана креатинкіназа – фермент, який каталізує передачу макроергічного фосфатного залишку АТФ на креатин з утворенням креатинфосфату. Останній дифундує у саркоплазму до міофібрил. Тут розчинна форма креатинкінази каталізує взаємодію креатинфосфату з АДФ, який утворився при скороченні – при цьому утворюється АТФ. Креатинкіназа складається із 2-х субодиниць (М та В), має 3 ізоферменти – ММ ( у мітохондріях), МВ та ВВ (обидва у саркоплазмі). МВ є лише у серцевому м’язі, ММ – переважно у скелетному м’язі, ВВ – здебільшого у мозку. Визначення цих ферментів має діагностичне значення. Утворення АТФ шляхом анаеробного гліколізу в серці не відіграє суттєвої ролі – це пояснює чутливість міокарду до недостачі кисню.

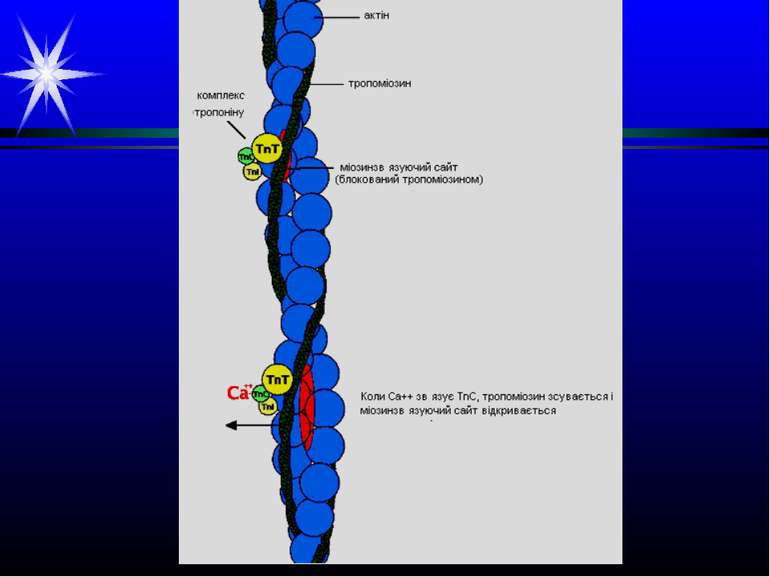
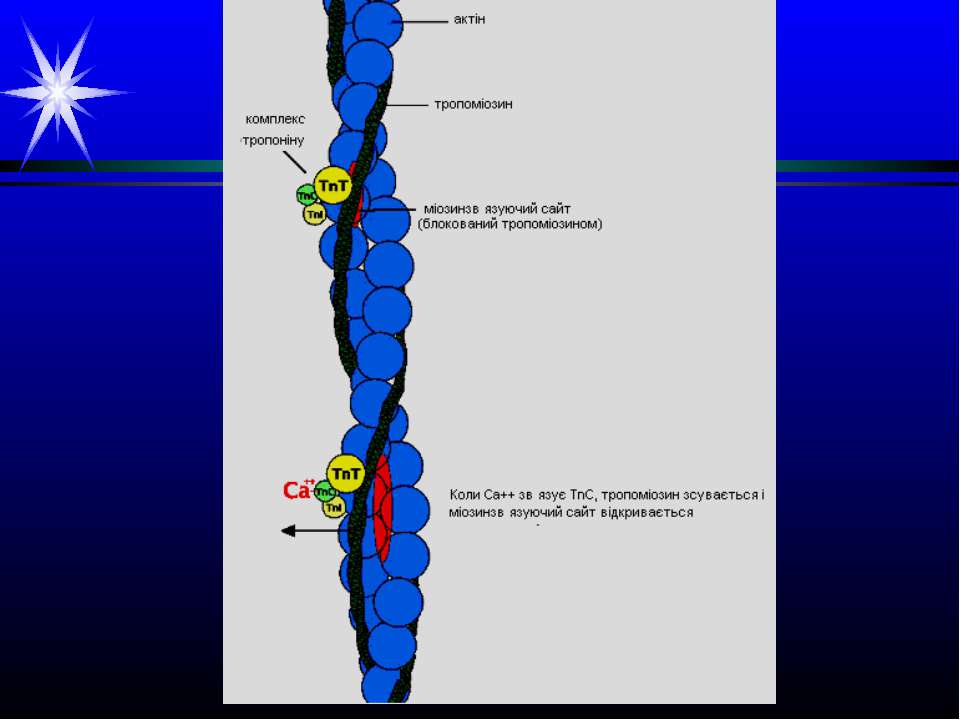
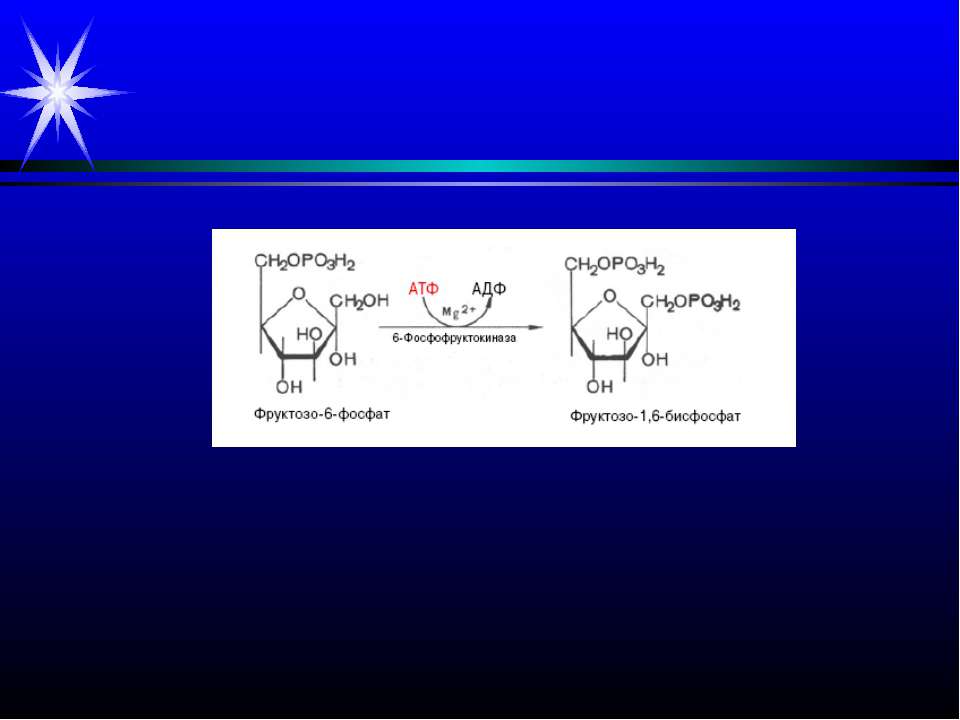
Патологія м’язової тканини. Інфаркт міокарда. Ішемія міокарда – зменшення його постачання кров’ю. Внаслідок розвивається гіпоксія, припиняється утворення АТФ шляхом окисного фосфорилювання. Зниження вмісту АТФ і зростання АМФ активують ключовий фермент гліколізу фосфофруктокіназу, яка включає інтенсивний розпад глікогену до молочної кислоти. Тобто якщо у нормі молочна кислота, яка надходить із скелетних м’язів через кров у міокард, перетворюється на піруват завдяки ферменту ЛДГ, а потім піруват підлягає окисному декарбоксилуванню у мітохондріях, то при ішемії утворюється надлишок молочної кислоти. Хоча при цьому також утворюється невелика кількість АТФ, збільшення лактату спричиняє розвиток ацидозу, і активність фосфофруктокінази вже гальмується, гліколіз глікогену і утворення при цьому АТФ припиняється. Нестача АТФ і закислення середовища порушують іонний баланс. Зменшується надходження у клітини Са++ через потенціалзалежні кальцієві канали; іони Са++ звільняються із комплексів з тропоніном, гальмується АТФазна активність міозину, руйнуються поперечні містки. Це зумовлює зниження скоротливості міокарду. При тривалій ішемії пошкоджуються мембрани саркоплазматичного ретикулуму, плазматичні мембрани, Са++ надходить у саркоплазму за градієнтом концентрації. В клітині зростає вміст Са++, Nа+, лактату, пірувату, продуктів розпаду АТФ – це зумовлює надходження у клітину рідини та її розбухання. Зростає інтенсивність ПОЛ мембран, їх проникність, у кров із клітин виходять ряд ферментів. Різко знижується вміст білків міофібрил, накопичуються білки строми. Вільні жирні к-ти не окислюються, а включаються у тригліцериди, що викликає жирову інфільтрацію серцевого м’язу.

Інфаркт міокарда: діагностика З метою діагностики інфаркту міокарда визначають у плазмі крові креатинкіназу, АсАТ, ізоферменти ЛДГ1 та ЛДГ2, які виходять у кров із міоцитів. - через 6 годин після інфаркту у крові є максимальним рівень МВ-креатинкінази, ця величина триває 12 годин. Надалі звільняється ММ-креатинкіназа. Сумарна креатикіназна активність плазми крові є максимальною через 24-48 годин, зберігається 3-5 днів. - АсАТ – зростає у 10-100 разів, максимум – через 1-2 дні, утримується 4-6 днів. - ЛДГ1 – максимум – через 2-3 дні; зберігається 7-12 днів. - Визначають активність глікогенфосфорилази та її ізоферменту ГФ-ВВ. Активність останнього зростає вже у перші 3-4 години після інфаркту. Протягом 48 годин повертається до норми. Маркером інфаркту є і визначення у крові вмісту міоглобіну. Норма – нижче 80 нг/нл. Протягом перших 2-х годин після інфаркту його вміст різко зростає. Ця величина також швидко повертається до норми, тому її визначення доцільне для діагностики повторних інфарктів ( тих, які розвиваються протягом доби). В той же час необхідна диференційна діагностика – адже зростання вмісту міоглобіну може спричинятися і ураженням скелетних м’язів. Вміст серцевих білків тропонінової системи – тропонін Т, тропонін І: вміст останнього зростає у 100 разів від верхньої межі норми.

Хронічні серцеві хвороби У розвитку хронічної серцевої недостатності велика роль належить зростанню нейрогормональної активності. На початку захворювання з метою підтримки гомеостазу у крові різко зростає вміст норадреналіну. Постійно підвищений його рівень стає шкідливим фактором: веде до некрозів у міокарді та ін. зсувів. З часом виділення норадреналіну нервовими закінченнями адренергічних нервів міокарду пригнічується, і компенсаторний механізм стає невдосконалим. Речовини, які інгібують вплив нейрогормонів, сповільнюють наростання хронічної серцевої недостатності.

Міокард і старіння Специфічними ознаками, що характеризують міокард в умовах старіння є: 1) Атрофія міокарду – генетично запрограмоване руйнування клітин міокарду; це спричиняє гіпертрофію міозитів, які залишилися, та гіпертрофію сполучної тканини міокарду. Ознака – гіпертрофія лівого шлуночка: потовщення його стінки. 2) зниження активності міозинової АТФази. 3) сповільнення синтезу білків, послаблення активності Са2+, Мg2+-АТФази мембран саркоплазматичного ретикулуму. 4) зниження здатності міокарду до поглинання кисню та максимального числа серцевих скорочень при навантаженнях.

Скелетні м’язи й старіння Специфічними ознаками, що характеризують скелетні м язи в умовах старіння є: 1) Зменшення вмісту АТФ, креатинфосфату у м’язах; зменшення вмісту глікогену, зростання вмісту жиру. Це спричиняє зниження здатності до продукування енергії в кожному поперечному містку між міозином і актином у м’язовому філаменті. 2) Зниження рН, зростання вмісту неорганічного фосфату у „старих” м’язах. 3) Зниження активності гліколітичних ферментів, у тому числі ЛДГ, міокінази.

М’язові дистрофії Специфічними ознаками, що характеризують м язові дистрофії є: 1) Зменшення вмісту міофібрилярних білків і зростання вмісту колагену та еластину; 2) Зниження АТФазної активності міозину, гліколітичних ферментів, зростання активності ферментів лізосом; 4) Внаслідок порушення обміну вуглеводів зменшується вміст АТФ і креатинфосфату. 5) Зміна фосфоліпідного складу мембран: зростання вмісту сфінгомієліну і лізофосфатидилхоліну, зменшення вмісту фосфатидилхоліну й фосфатидилетаноламіну. 6) Характерна ознака - порушення метаболізму креатину. В нормі у людини щоденно синтезується 1-2 г креатину; до 150 мг його виводиться із сечею у незмінному вигляді, більшість (18-32 мг/ кг маси тіла у чоловіків, 10-25 мг/кг у жінок) – у формі креатиніну, що утворюється при неферментативному дефосфорилюванні креатинфосфату. При м’язових дистрофіях утворення і виведення креатиніну знижується, а вміст креатину у сечі – зростає (креатинурія). 7) Діагностична ознака – зростання активності у плазмі крові ферментів, характерних для м’язів – креатинкінази ( в 10 разів на ранніх стадіях; пізніше – знижується до норми) та амінотрансфераз.

Види м’язових дистрофій Найпоширеніші - Дюшена, Беккера, Емеві-Драйфуса. М’язова дистрофія Дюшена – спадкове захворювання, закінчується летально. В основі – мутація гену Х-хромосоми (хворіють хлопчики), що кодує білок цитоскелету дистрофін, який надає міцності плазматичній мембрані м’язових клітин. За відсутності цього білку плазматична мембрана міоциту руйнується під час скорочення м’язів, що веде до повної загибелі клітин. Дистрофія Беккера – кількість дистрофіну різко зменшена і змінена його молекулярна структура

Метаболічні міопатії В основі – порушення обміну речовин у м’язовій тканині. Первинні (спадкові) – дефекти метаболізму вуглеводів, ліпідів, пуринів, порушення циклу Кребса, клітинного дихання, розподілу кальцію та електролітів. Вторинні (набуті) – в основі мають ендокринні хвороби, порушення харчування і обміну електролітів. 1) міопатії, пов’язані з порушенням обміну глікогену – через відсутність ферменту фосфорилази. Джерелом енергії є вільні жирні кислоти, хворі не виявляють м’язової втоми при легких фізичних вправах. 2) міопатії, пов’язані з накопиченням жирних кислот – спричиняє накопичення ліпідів між міофібрилами та зниження витривалості м’язів до фізичного навантаження. 3) мітохондріальні міопатії – змінюється вміст мітохондрій у міоцитах, їх розмір та внутрішня структура. Це спричиняє роз’єднання окисного фосфорилювання і дихання у компонентах дихального ланцюга. Супроводжується м’язовою слабкістю. 4) міопатії, пов’язані з недостатністю карнітину. Джерелом карнітину є молочні продукти та червоне м’ясо, він може синтезуватися з лізину або метіоніну в печінці, мозку, нирках. У м’язах – 98% запасу цієї сполуки. Карнітин бере участь у транспорті вільних жирних кислот із довгим вуглецевим ланцюгом усередину мітохондрій, де вони окислюються. У хворих знижується вміст карнітину у скелетних м’язах, серцевому м’язі, печінці, у плазмі крові. Спричиняє прогресуючу м’язову слабкість. 5) міопатії, пов’язані з порушенням пуринового обміну – недостатність міоаденілатдезамінази – ферменту, що каталізує перетворення АМФ на інозинмонофосфат. Спричиняє швидку втомлюваність, біль у м’язах.
Схожі презентації











Категорії