Презентація на тему:
P-gl_Eng Lecture
Завантажити презентацію




 (SLC) - Primar...")
 Pharmacokinetic – Absorption, metabolism, excretion of drugs 1. Located in...")
 Pharmacodynamics – Target for many drugs - SERT (SLC6A4), serotonin transp...")




 Epithelial cells of gastrointestinal tract Influx of essential nutrients a...")

 Hepatocytes in liver Even if drug is absorbed it is expelled into bile, en...")
 Renal epithelial cells Efflux of drugs into tubular lumen, easily excreted")
 Tissues with barrier function Blood brain Blood testis Blood placenta limi...")
 Cancer cells Highly expressed, leading to efflux of anticancer agents from...")















P-gl_Eng Lecture
Завантажити презентаціюПрезентація по слайдам:

Dr. Ganna Tolstanova, Associated Professor of Dep. of Biochemistry, ESC “Institute of Biology”

Introduction Structure Location and functions Substrates and inhibitors Important interactions Future trends Conclusion

Transporters are membrane proteins Control the influx of essential nutrients and ions and the efflux of cellular waste, toxins and drugs Controls cellular homeostasis
 (SLC) - Primar...")
Transporters in body ATP binding cassette Solute carrier (ABC) (SLC) - Primary active transporters (rely on ATP hydrolysis) 49 known genes for ABC proteins (7 families - ABCA to ABCG) P-glycoprotein (P-gp, encoded by ABCB1, also termed MDR1) - Facilitated transporters and ion-coupled secondary active transporters (antiport) - 300 transporters (43 families – SLC1-SLC43) Drug targets, drug absorption and disposition SERT (serotonin transporter, SLC6A4) DAT (dopamine transporter, SLC6A3)
 Pharmacokinetic – Absorption, metabolism, excretion of drugs 1. Located in...")
1) Pharmacokinetic – Absorption, metabolism, excretion of drugs 1. Located in intestinal, renal, and hepatic epithelium 2. Selective absorption and elimination of endogenous substances and xenobiotics, including drugs 3. Work in concert with drug metabolizing enzymes 4. Tissue specific drug distribution 5. Protective barriers to particular organs and cell types
 Pharmacodynamics – Target for many drugs - SERT (SLC6A4), serotonin transp...")
2) Pharmacodynamics – Target for many drugs - SERT (SLC6A4), serotonin transporter - DAT (SLC6A3), dopamine transporter 3) Drug resistance - Anticancer drugs (BCRP (ABCG2)) - Antiviral agents (MRP4 (ABCC4))

- Most important member of ABC family - Transmembrane protein coded by MDR-1 gene - Functions to actively pump a diverse array of xenobiotics out of the cells in which it is expressed - Display a high level of constitutive (basal) ATPase activity, which is observed in the absence of added drugs

First described in 1976, by JULIANO & LING in Chinese hamster ovary cells selected in culture for colchicines resistance PULLIAM et al. in 1985 discovered the particular increased penetration of ivermectin through the BBB resulting in 31-fold higher concentration of the drug in the brain than in the plasma in MDR knocked out mice

MICKISCH et al., 1991, found P-glycoprotein inhibition due to cyclosporine enhanced paclitaxel (Taxol) oral bioavailability

Single polypeptide Two homologous halves Each contains six transmembrane domains and a hydrophilic region with an ATP-binding domain
 Epithelial cells of gastrointestinal tract Influx of essential nutrients a...")
1) Epithelial cells of gastrointestinal tract Influx of essential nutrients and ions and efflux of cellular waste , drugs etc. Extensive overlap in the substrate specificities and tissue localization of P-gp and drug metabolizing enzyme in the cytochrome P450 (CYP3A4)

Lead to the hypothesis that these two proteins work together to protect the body from absorption of harmful xenobiotics, acting synergistically in the small intestine
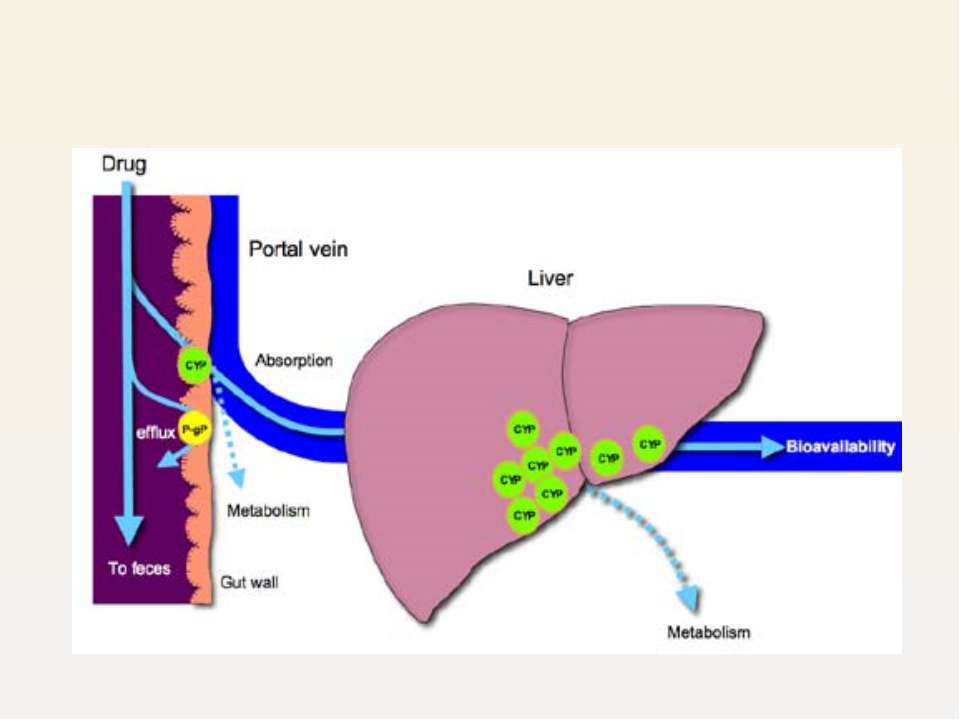
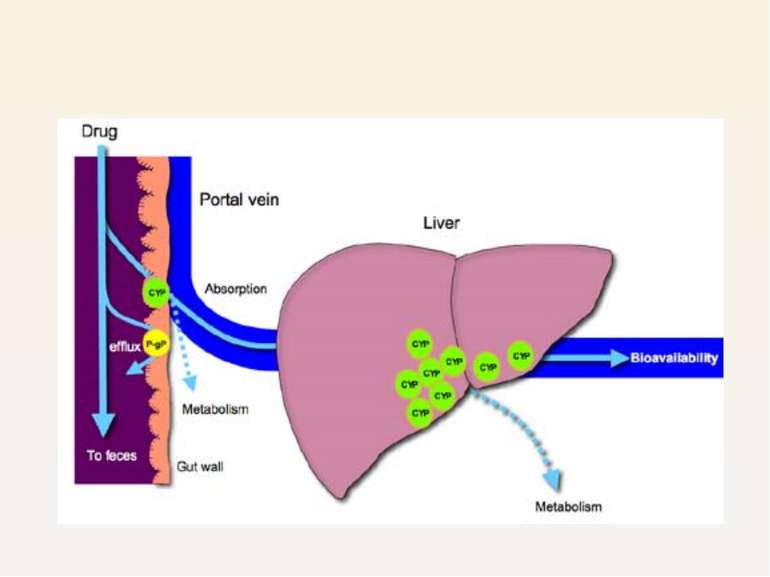
 Hepatocytes in liver Even if drug is absorbed it is expelled into bile, en...")
2) Hepatocytes in liver Even if drug is absorbed it is expelled into bile, enters intestine again Work synergistically with CYP3A4 in liver
 Renal epithelial cells Efflux of drugs into tubular lumen, easily excreted")
3) Renal epithelial cells Efflux of drugs into tubular lumen, easily excreted
 Tissues with barrier function Blood brain Blood testis Blood placenta limi...")
4) Tissues with barrier function Blood brain Blood testis Blood placenta limiting entry and accumulation of many drugs into the brain and contributing to the protective function of the central nervous system
 Cancer cells Highly expressed, leading to efflux of anticancer agents from...")
5) Cancer cells Highly expressed, leading to efflux of anticancer agents from cells Resistance to these agents

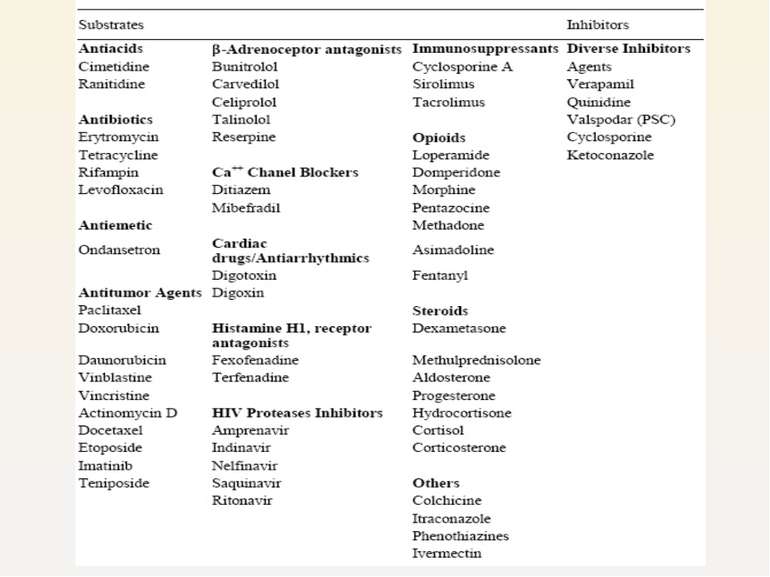
Various classes of drugs Resulting in efflux of drug from body, lowering their plasma concentration
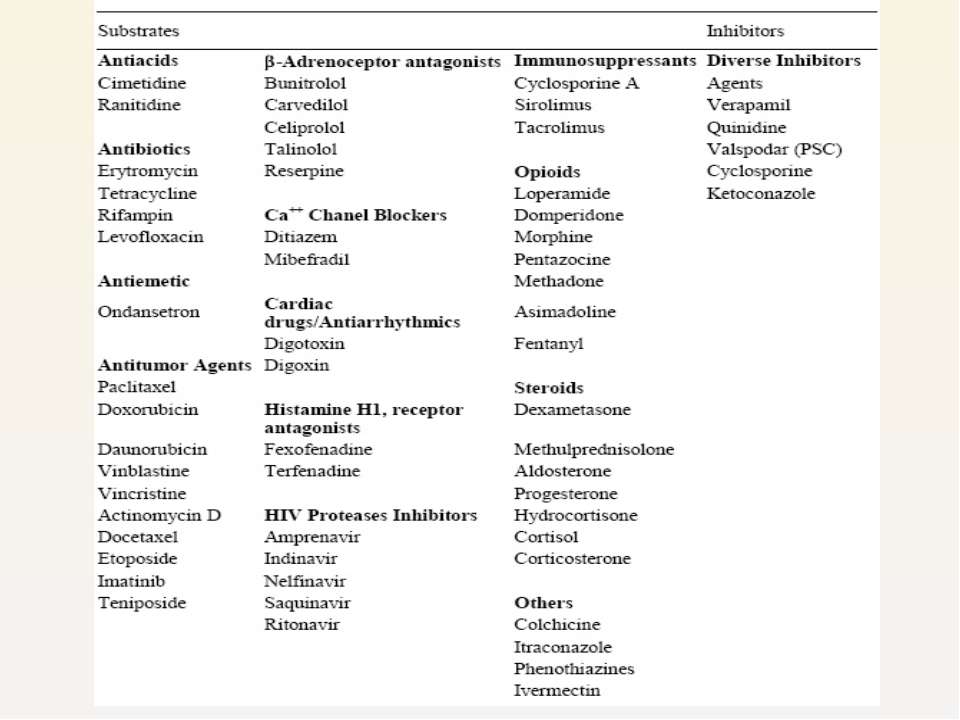

P-gp induction or inhibition may have a substantial effect on the pharmacokinetics and pharmacodynamics of concomitantly administered drugs that are substrates for this transporter.

Digoxin Absorption reduced due to co-administration of rifampicin (MDR1 inducer) Renal excretion retarded due to concurrent administration of MDR1 inhibitors (verapamil, quinidine etc.)

Loperamide Peripheral opioid for diarrhea, substrate for P-glycoprotein Co-administration of quinidine (MDR1 inhibitors) has caused marked respiratory depression

Protease inhibitors Ritonavir, Nelfinavir, Indinavir, Saquinavir substrates for P-glycoprotein P-gp transport at the intestinal and/or hepatic level limits the systemic bioavailability Poor penetration into the brain, testis Treatment ineffective and development of resistance faster

Anticancer agents MDR1 highly expressed in cancer cells, rendering resistant to anticancer agents Reduced bioavailability Paclitaxel, actinomycin D, vincristine etc. substrates for P-gp.

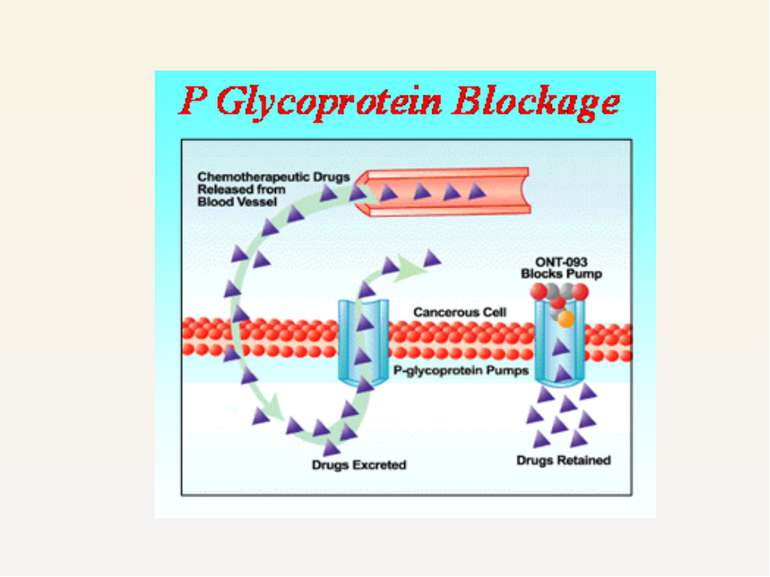
P-gp induction does not have a therapeutic role till date, P-gp inhibition is an attractive therapeutic approach to reverse multidrug resistance

Strategies to modulate P-gp function are being pursued actively in oncology reversal of multidrug resistance in tumours several newly developed agents that inhibit P-gp function are in phase I-III clinical trials. e.g. ONT 093 PSC 31665
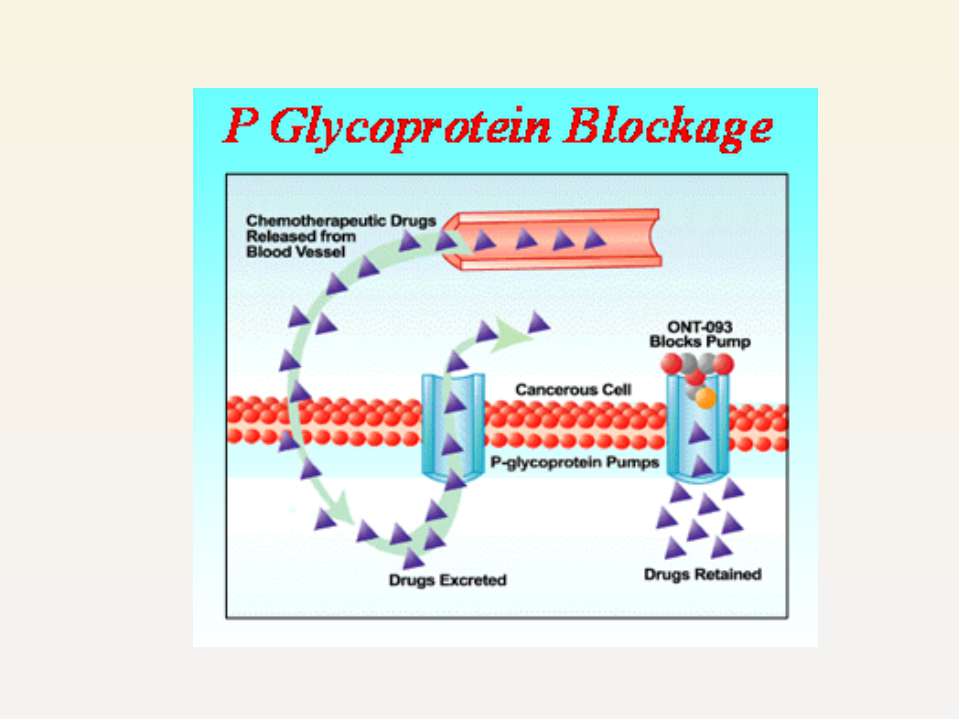

Blockage of P-gp may be useful in facilitating greater intestinal absorption, bioavailability and penetration of protease inhibitors into HIV sanctuary sites as well as reduced excretion. Simplify protease inhibitor-containing regimens by reducing the oral doses of protease inhibitors and the frequency at which they are taken

MDR1 gene polymorphisms increases susceptibility to diseases such as Parkinson's disease, inflammatory bowel disease, multi-drug resistant epilepsy and renal carcinoma. MDR1 induction beneficial ???

P-glycoprotein encoded by MDR1 gene that influences drug pharmacokinetic and Pharmacodynamics P-gp inhibition is an attractive therapeutic approach to reverse multidrug resistance

Kathleen M. Giacomini and Yuichi Sugiyama membrane transporters and drug response ; Goodman and Gilman's, Pharmacological basis of therapeutics;11;40-65;2005. Bertram G. Katzung, Introduction, Basic and clinical pharmacology:11:9-10;2009

BRADY, J.M. et al., Tissue distribution and chemical induction of multiple drug resistance genes in rats; Drug Metabolism and Disposition, Baltimore; vol.30; 7; 2005. CUMMINS, C. L. et al. In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: Studies using the rat single-pass intestinal perfusion model; Journal of Pharmacology and Experimental Therapeutics, Baltimore; v.305, n.1; p.306-314; 2006.

FROMM, M.F. Importance of P-glycoprotein at blood-tissue barriers; TRENDS in Pharmacological Sciences; v.25; n.8; p.423-429; 2004. KHARASCH, E.D. et al. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. British Journal of Clinical Pharmacology, Oxford, v.57; n.5; p.600-610, 2003.

THANK YOU
Схожі презентації

Категорії