Презентація на тему:
предмет та завдання фармакокінетики. Взаємозв’язок будови клітини і фармакокінетичних параметрів
Завантажити презентацію









 - це її кількість у певному об'ємі кров...")
 - (уявний гіпотетичний об'єм розподілу лікарської речови...")

 - площа фігури...")
 визначають відносною кількістю лікарської речовини, що над...")

 - це співвідношення кількості...")

 - це умовний об'єм крові чи ЇЇ плазми, що звільняється...")

 Це фармакокінетичний п...")
 - час, необхідний для всмоктування половини доз...")
 - відсоток зменшення концентрації лікарської речов...")
















 ліка...")


 - основна стру...")

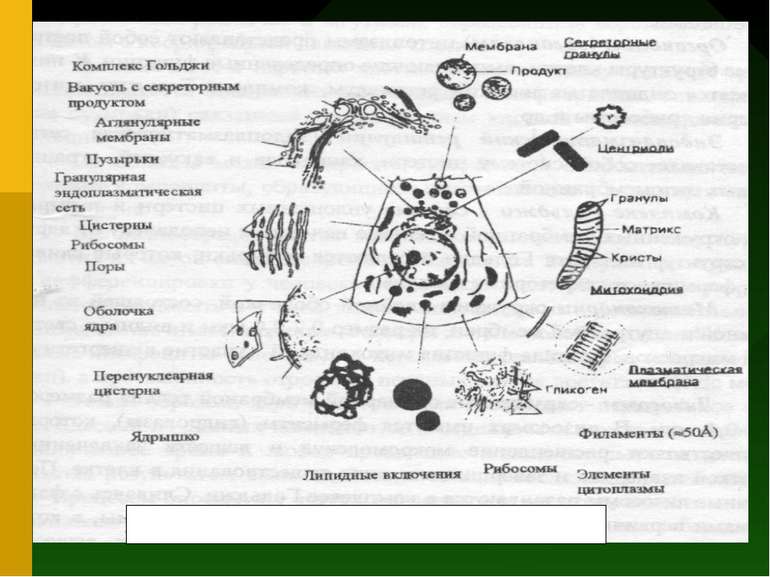
 цитоплазми являють собою постійні структури клітини, що ...")




предмет та завдання фармакокінетики. Взаємозв’язок будови клітини і фармакокінетичних параметрів
Завантажити презентаціюПрезентація по слайдам:

Тема: Вступ: предмет та завдання фармакокінетики. Взаємозв’язок будови клітини і фармакокінетичних параметрів

Фармацевтична фаза Фармакокінетична фаза Фармакодинамічна фаза Послідовність фаз взаємодії лікарської речовини з організмом

Фармакокінетика (грец. pharmakon - лікарський засіб, отрута, зілля і kinetikos - те, що стосується руху) - розділ фармакології, який вивчає: надходження (шляхи введення); транспорт (абсорбція, всмоктування); розподіл, перетворення (біотрансформація) лікарських речовин в організмі; виведення (екскреція, елімінація) їх з організму, а також ефективність і переносимість препаратів залежно від цих процесів.

Введення лікарської речовини Всмоктування Вільна ЛР в плазмі Зв’язана з білками ЛР Ефект Розподіл в неактивних тканинах Ниркова екскреція Виведення в незмінному вигляді Реабсорбція Біотрансформація в метаболіти Виведення метаболітів Кров Загальна схема фармакокінетичних процесів лікарських засобів

Розрізняють клінічну й експериментальну фармакокінетику. Клінічна фармакокінетика досліджує процеси надходження, розподілу, біотрансформації та екскреції лікарських речовин, а також виявлення зв'язків між концентрацією лікарської речовини і (чи) ЇЇ метаболітів у біологічних рідинах і тканинах та фармакологічним ефектом. Клінічна фармакокінетика визначає таке дозування лікарських речовин, яке забезпечує їх необхідну концентрацію в середовищах організму для досягнення оптимального лікувального ефекту. Головне завдання експериментальної фармакокінетики - вивчення трансформації лікарських засобів в організмі тварин у нормі і при моделюванні різних захворювань.

Для визначення фармакокінетичних показників організм людини чи експериментальної тварини розглядається як особливе біологічне середовище, де відбувається розподіл лікарських засобів в органах, тканинах, клітинах, субклітинних структурах, біотрансформація, а також взаємодія лікарських речовин з тканинними рецепторами.

Одним з основних показників, що визначають ефективність лікарських засобів, є концентрація лікарської речовини в ділянці рецептора або тканини, де відбувається їх взаємодія. Визначити концентрацію лікарських речовин в організмі людини, в окремому органі чи тканині практично неможливо. Для визначення фармакокінетичних параметрів реєструють кількість лікарської речовини в крові, оскільки здебільшого існує залежність між концентрацією речовини в крові і в ділянці рецептора. Ґрунтуючись на отриманих даних, будують графік - фармакокінетичну криву. На осі ординат зазна чають концентрацію речовини у плазмі кро ві, а на осі абсцис - термін дослідження.


ГОЛОВНІ ПОНЯТТЯ ФАРМАКОКІНЕТИКИ Камера - умовне поняття в фармакокінетиці, під яким розуміють простір, що має певний об'єм і концентрацію лікарської речовини в цьому просторі. Це не є анатомічний простір. Це одиниця формалізованої фармакокінетичної системи для математичного моделювання процесів взаємодії лікарської речовини з організмом. Розрізняють центральну камеру - кров і органи, що мають інтенсивне кро вопостачання (серце, нирки, легені, ендокринні залози, печінка, кишки), і периферичну - органи з менш інтенсивним кровопостачанням (шкіра, підшкірна клітковина, м'язи, жирова тканина та ін.). Умовно взаємодія лікарської речовини з організмом відбувається за однокамерною або багатокамерною моделлю і характеризується концентрацією лікарської речовини (Сл) та об'ємом розподілу (Vd).
 - це її кількість у певному об'ємі кров...")
Концентрація лікарської речовини (Сл) - це її кількість у певному об'ємі крові в конкретний момент після введення в організм. Концентрацію лікарської речовини визначають спектрофотометричним, хроматографічним, ферментативним, радіоімунним та іншими методами і виражають у міліграмах на 1 л, у мікрограмах на 1 мл або у відсотках. Динаміка концентрації лікарської речовини в організмі залежить від шляхів уведення, дози, фізико-хімічних властивос тей, тривалості дії тощо. Найпростішою фармакокінетичною моделлю є однокамерна, коли організм вважається єдиною гомогенною камерою. Однокамерну модель застосовують для визначення концентрації лікарських речовин у крові, плазмі та сироватці крові, а також у сечі. Фармакокінетичні процеси найбільшою мірою відповідають процесам у цілому організмі за дво- чи трикамерною моделлю.
 - (уявний гіпотетичний об'єм розподілу лікарської речови...")
Об'єм розподілу (Vd) - (уявний гіпотетичний об'єм розподілу лікарської речовини) - це умовний об'єм рідини, потрібний для рівномірного розподілу введеної дози лікарської речовини до концентрації, що визначається у крові в момент дослідження Vd=D/C0 Визначається у літрах (л). Умовний об'єм розподілу – це об'єм віднесений до маси тіла (літрів на 1кг маси тіла - л/кг). Об'єм розподілу лікарської речовини певною мірою визначає ступінь проникнен ня її з крові й позаклітинної рідини у тка нини, а також створення її депо в органах. Наприклад, для антибіотиків-аміноглікозидів, які мало розчинні в ліпідах, об'єм розподілу близький до об'єму позаклітинної рідини, а для добре розчинних у ліпідах тетрациклінів - він є значно вищим. Об'єм розподілу залежить від шляху введення, дози, фізико-хімічних властиво стей лікарської речовини (розчинність у ліпідах і воді, ступінь йонізації й поляр ності, молекулярна маса), а також віку, статі хворого, кількості рідини в організмі, пато логічного стану (захворювання печінки, нирок, серцево-судинної системи, кишок). Однокамерну модель можна викорис товувати для визначення концентрації лі карської речовини у таких середовищах організму, як кров (або її сироватка, плаз ма) і сеча. Сечу досліджують щодо речо вин, здатних швидко розподілятися в різ них середовищах (рідини, тканини) орга нізму. Суть цієї моделі полягає в тому, що вона передбачає однотипну динаміку змін концентрації речовини у плазмі кро ві й тканинах, а також швидше встанов лення рівноваги між процесами надход ження і виведення її з організму.

Достовірніші результати отримують вивченням фармакокінетичних процесів за дво- чи трикамерною моделлю. При цьому плазму крові, а також печінку, нирки, легені, серце та інші органи з інтенсивним кровопостачанням вважають центральною, або меншою, камерою, а шкіру, жирову і м'язову тканини, рівень кровопостачання яких менший, - периферичною, або більшою. Речовини в меншій камері розподіляються швидко, а у великій - повільно. У міру виведення речовини з організму її концентрація знижується. Цей період позначають як фазу перерозподілу ( -фаза). У подальшому виведення лікарської речовини прискорюється, відбувається міграція її з великої камери до меншої, що позначають як фазу виведення ( -фаза). Між - і -фазами у певному часовому інтервалі настає стан рівноваги.
 - площа фігури...")
Площа під кривою "концентрація - час" (AUC - area under curve) - площа фігури, обмежена фармакологічною кривою й осями координат (AUC= С0/Kel ); Площа під кривою залежить від об'єму розподілу і загального кліренсу; Площа під фармакокінетичною кривою “концентрація-час” у разі лінійної залежності є пропорційною кількості лікарської речовини, що міститься у крові.
 визначають відносною кількістю лікарської речовини, що над...")
Біодоступність (F) визначають відносною кількістю лікарської речовини, що надходить до загального кола кровообігу і взаємодіє з тканинними рецепторами. Біодоступність виражають у відсотках і визначають за формулою: Однакові дози Різні дози

Біодоступність залежить від хімічної будови речовини, технології виготовлення лікарської форми, ступеня абсорбції її у кров з травного каналу при ентеральному введенні, біотрансформації при першому пасажі крізь печінку, швидкості транспорту при парентеральному введенні. Біодоступність лікарської речовини виражають у відсотках і при введенні безпосередньо в кров приймають за 100 %.
 - це співвідношення кількості...")
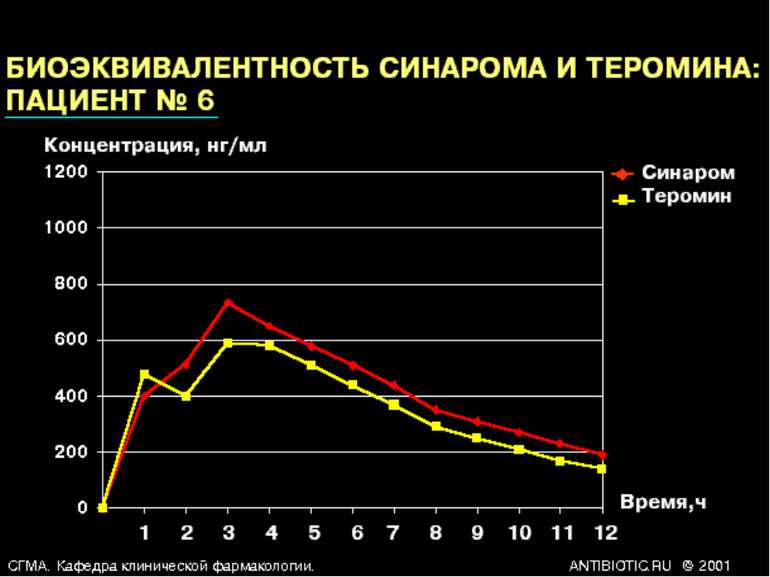
Біоеквівалентність (порівняльна біодоступність) - це співвідношення кількості лікарської речовини, що надходить у кров при введенні її в різних лікарських формах (або лікарських препаратів різних фірм). Вивчення біоеквівалентності дає змогу порівнювати ефективність різ них лікарських препаратів.

Біофаза - ділянка безпосередньої взаємодії лікарської речовини з рецептором або тканинною структурою, в тому числі клітинною оболонкою, мітохондрійною, ендоплазматичною, лізосомною, рибосомною мембранами.
 - це умовний об'єм крові чи ЇЇ плазми, що звільняється...")
Загальний кліренс (Сl) - це умовний об'єм крові чи ЇЇ плазми, що звільняється (очищається) від лікарської речовини за одиницю часу. Виражають у літрах чи мілілітрах за 1 хв і розраховують за формулою: де Сl - загальний кліренс; D - доза вве деного препарату; AUC — площа під фармакокінетичною кривою. Розрізняють нирковий кліренс - швидкість виведення лікарської речовини з сечею, печінковий - швидкість її інактивації в печінці, і жовчний - швидкість виведен ня лікарської речовини з жовчю. Нирковий кліренс (С1r) відбиває елімінацію лікарської речовини з організму і є співвідношенням кількості лікарської речо вини, що екскретується з сечею, і концент рації її у плазмі крові: де Сu - концентрація лікарської речовини в сечі, мг/мл; Ср - концентрація лікарсь кої речовини у плазмі крові, мг/мл; V -швидкість виділення сечі, мл/хв.

Для визначення кліренсу застосовують лікарську речовину, яка не метаболізується і повністю виводиться з організму в незміненому вигляді. У таких випадках значення кліренсу відбиває функціональну актив ність органів виділення. За нормальної функції органів виділення, зокрема в дослідженнях на експериментальних тваринах, рівень кліренсу незміненої лікарської речовини свідчить про ступінь його метаболічних перетворень в організмі.
 Це фармакокінетичний п...")
Період напіввиведення - (період напівелімінації, Т1/2) Це фармакокінетичний показник часу, протягом якого кіль кість лікарської речовини в камері або його концентрація в крові зменшується на 50 %. де Т1/2 - період напіввиведення; 0,693 -коефіцієнт, що є логарифмом із 2; Vd -об'єм розподілу; СІ - загальний кліренс. Або ж Т1/2 = 0,693/Kel Наведена формула свідчить, що Т1/2 не може повністю відобразити процес виді лення лікарської речовини з організму. Це зумовлено тим, що за певних умов (зменшення кровотоку в органах, особливо в нирках, недостатнє проникнення крізь мембрани) можливе зменшення розподі лу кліренсу лікарської речовини, внаслідок чого Т1/2 суттєво не змінюється, і це може призвести до накопичення в крові токсичних концентрацій призначеної лікарської речовини.
 - час, необхідний для всмоктування половини доз...")
Період напівабсорбції (T1/2a) - час, необхідний для всмоктування половини дози лікарського препарату з місця введення в системний кровоплин; пропорційний швидкості абсорбції Т1/2а= 0,693/Ка Період напіврозподілу (Т1/2р) - час, необхідний для досягнення концентрації препарату в крові, рівної 50% від рівноважної; тобто при наявності рівноваги між кров'ю і тканинами,
 - відсоток зменшення концентрації лікарської речов...")
Константа елімінації (Кel) - відсоток зменшення концентрації лікарської речовини в крові за одиницю часу. Чим більша Кel, тим швидше лікарська речовина видаляється з крові. Якщо, наприклад, Кel, двох лікарських речовин становлять 0,25 год-1 і 0,025 год-1, це означає, що щогодини концентрація в крові цих речовин зменшується відповідно на 25 і 2,5 % одноразової початкової концентрації. Константи швидкості абсорбції (всмоктування) (Ка) і екскреції (виведення) (Кех) - характеризують відповідно швидкість зникнення лікарського препарату з організму шляхом метаболізму і виведення, швидкість надходження його з місця введення в кров і швидкість екскреції з сечею, калом, слиною й ін.

Фармакокінетичний процес можна уявити у вигляді таких взаємопов'язаних етапів: 1. Введення лікарської речовини в організм. 2. Вивільнення лікарської речовини з лікарської форми. 3. Абсорбція лікарської речовини - проникнення крізь біологічні мембрани в судинне русло і тканини до специфічного клітинного рецептора. 4. Розподіл лікарської речовини в біологічних рідинах, органах д тканинах. 5. Біотрансформація (перетворення) лікарської речовини - біохімічне перетворення (метаболізм) із зміною фармаколо гічних властивостей і утворенням метабо літів, які виводяться з організму. 6. Виведення (екскреція, елімінація) лікарської речовини або її метаболітів з організму.

Всмоктування лікарських засобів Всмоктування лікарських засобів — це процес надходження ЛЗ із місця введення в кров. Всмоктування залежить від шляху введення, розчинності лікарського засобу в тканинах у місці його введення і кровотоку в цих тканинах. Швидкість проходження більшості лікарських препаратів через слизову оболонку травного тракту визначається їхньою розчинністю в ліпідах і іонізацією. Деякі лікарські засоби всмоктуються шляхом активного транспорту. При прийомі ЛЗ усередину варто враховувати, що швидкість їхньої абсорбції в різних відділах шлунково-кишкового тракту (ЖКТ) неоднакова. Після проходження через стінку шлунка і/чи кишечнику лікарський препарат надходить у печінку. Деякі ЛЗ під впливом ферментів печінки піддаються значним змінам («ефект первинного проходження»). Саме тому, а не внаслідок поганої абсорбції, щоб досягти ефекту, дози деяких препаратів (пропранололу, аміназину, опіатів) при прийомі їхній усередину повинні бути значно більше, ніж при внутрішньовенному введенні. Біотрансформацію речовини при первинному проходженні через печінку в процесі всмоктування називають пресистемним метаболізмом. Інтенсивність пресистемного метаболізму залежить від швидкості крові в печінці.

На процес всмоктування лік у шлунку і кишечнику впливає рН, що у шлунку дорівнює 1—3, у дванадцятипалій кишці — 5—6, а в тонкій і товстій кишках — приблизно 8. Кислоти легше всмоктуються в шлунку, підстави — у тонкій і товстій кишках. Під дією кислого середовища шлунка деякі лікарські засоби, зокрема бензилпеніцилін, можуть руйнуватися. На лікарські препарати діють і ферменти ШКТ, що здатні ін активувати їх і поліпептиди: адренокортикотропний гормон (АКТГ), вазопресин, інсулін і т.д., а також деякі інші речовини: прогестерон, тестостерон, альдостерон. Солі жовчних кислот можуть прискорити всмоктування лікарських засобів чи сповільнити його при утворенні нерозчинних сполук. На всмоктування ЛЗ впливають також моторика ШКТ, об’єм і склад їжі, кількість прийнятої рідини, інтервал часу між їжею і прийомом препаратів. Так молоко порушує всмоктування тетрациклінів, ампіциліну й амоксициліну. Варто враховувати і стимулюючу дію їжі на секрецію шлункового соку і соляної кислоти.

Розподіл ЛЗ в організмі Після поступлення в системний кровоток лікарські засоби розподіляються по тканинах організму. Характер розподілу лікарського засобу визначається розчинністю його в ліпідах, ступенем зв'язування з білками плазми крові, інтенсивністю регіонарного кровотоку й іншими факторами. Велика частина ЛЗ у перші хвилини після всмоктування попадають у ті органи і тканини, що найбільш активно кровопостачаються: серце, печінка, нирки. Повільніше відбувається насичення лікарським препаратом м'язів, слизових оболонок, шкіри і жирової тканини. Для досягнення терапевтичних концентрацій ЛЗ у цих тканинах потрібно від декількох хвилин до декількох годин. Важливим фактором, що визначає розподіл ЛЗ, є швидкість його дифузії в різні тканини. Легко і швидко відбувається дифузія в інтерстиціальну тканину. Капіляри добре проникні і для водорозчинних, і для жиророзчинних речовин, тому водорозчинні препарати (наприклад, стрептоміцин), що погано всмоктуються з кишечнику, уводять парентерально. Вони добре проникають у позаклітинні області, але не діють на центральну нервову систему (ЦНС) і інші органи, потрапити в який речовина може тільки переборовши мембранні бар'єри. Розчинні в жирах препарати (наприклад, газоподібні анестетики) швидко розподіляються по всьому організмі, однаково добре проникаючи в позаклітинні і внутрішньоклітинні області.

Зв'язування ЛЗ із білками крові і тканин. Багато лікарських засобів володіють вираженою фізико-мімічною спорідненістю до різних білків плазми крові, насамперед до альбуміну. Зв'язування ЛЗ із білками плазми приводить до зниження їхньої концентрації в тканинах і в місці дії, тому що тільки вільний (незв'язаний) препарат проходить через мембрани. Речовина, що знаходиться в комплексі з білком, позбавлена специфічної активності. Вільна і зв'язана частини лікарського засобу знаходяться в стані динамічної рівноваги. Іноді ЛЗ накопичується в тканинах у значно великих концентраціях, чого можна чекати, виходячи з дифузійної рівноваги. Цей ефект залежить від градієнта рН, зв'язування лікарського засобу з внутрішньоклітинними елементами і його розподілу в жировій тканині. Клінічне значення мають випадки, коли з білками крові зв'язується більше 90 % ЛЗ.

Порушення зв'язування ЛЗ спостерігається при зниженні концентрації альбумінів у крові (гіпоальбумінемія) і здатності білків крові до з’єднання при деяких захворюваннях печінки і нирок. Навіть зниження рівня альбумінів у крові до 30 г/л (у нормі 33—55 г/л) може привести до значного підвищення вмісту вільної фракції фенітоїну. Клінічно значиме збільшення рівня вільної фракції фуросеміду відбувається при зниженні кількості альбуміну до 20 г/л.

Біотрансформація ЛЗ Під біотрансформацією, чи метаболізмом, розуміють комплекс фізико-хімічних і біохімічних перетворень лікарських засобів, у процесі яких утворяться полярні водорозчинні речовини (метаболіти), що легше виводяться з організму. У більшості випадків метаболіти лікарських засобів менш біологічно активні і менш токсичні, чим вихідні сполуки. Однак біотрансформація деяких речовин приводить до утворення метаболітів, більш активних у порівнянні з уведенням в організм речовинами.

Розрізняють два типи реакцій метаболізму лікарських препаратів в організмі: несинтетичні і синтетичні. Несинтетичні реакції метаболізму лікарських препаратів можна розділити на двох груп: ті, що каталізуються ферментами ендоплазматичного ретикулуму (мікросомні) і ті, що каталізуються ферментами іншої локалізації (немікросомні). До несинтетичних реакцій відносяться окислення, відновлення і гідроліз. В основі синтетичних реакцій лежить кон'югація лікарських засобів з ендогенними субстратами (глюкуронова кислота, сульфати, гліцин, глутатіон, метильні групи і вода). Сполучення цих речовин з лікарськими препаратами відбувається через ряд функціональних груп: гідроксильну, карбоксильну, амінну, епоксидну. Після завершення реакції молекула препарату стає більш полярної й отже, легше виводиться з організму.

Усі лікарські засоби, що вводяться усередину, до надходження в системний кровоток проходять через печінку, тому їх розділяють на дві групи: перша — з високим, друга — з низьким печінковими кліренсами. Для ЛЗ першої групи характерний високий ступінь екстракції гепатоцитами з крові. Здатність печінки метаболізувати ці препарати залежить від швидкості кровотоку. Печінковий кліренс ЛЗ другої групи залежить не від швидкості кровотоку, а від ємності ферментативних систем печінки, які метаболізують дані препарати. Останні можуть мати високий (дифенін, хінідин, толбутамід) чи низький ступінь зв'язування з білками (теофілін, парацетамол). Метаболізм речовин з низьким печінковим кліренсом і високою здатністю до зв'язування з білками залежить насамперед від швидкості їхнього зв'язування з білками, а не від швидкості кровотоку в печінці.

На біотрансформацію лікарських засобів в організмі впливають вік, стать, навколишнє середовище, характер харчування, захворювання і т.д. Печінка є основним органом метаболізму ЛЗ, тому будь-який її патологічний стан відбивається на фармакокінетиці препаратів. При цирозах печінки порушується не тільки функція гепатоцитів, але і її кровообіг. При цьому особливо змінюються фармакокінетика і біодоступність препаратів з високим печінковим кліренсом. Збільшення біодоступності лікарських засобів з високим печінковим кліренсом при пероральному застосуванні хворими цирозом печінки відбувається, з одного боку, зниженням метаболізму, з іншого боку — наявністю портокавальних анастомозів, по яких препарат надходить у системний кровообіг, минаючи печінку. Метаболізм препаратів з високим печінковим кліренсом, уведених внутрівенно, знижений у хворих цирозом печінки, однак ступінь такого зниження дуже різний. Коливання цього параметра залежить швидше за усе від здатності гепатоцитів метаболізувати лікарські засоби в залежності від характеру кровотоку в печінці.

Мікросомна біотрансформація У гепатоцитах найбільше повно представлений набір ферментних систем, що здійснюють окислення різноманітних ксенобіотиків (грец. xenos — чужий, bios — життя), тобто речовин, сторонніх для організму людини. До їхнього числа відноситься більшість лікарських засобів. Мікросомальному перетворенню піддаються насамперед жиророзчинні речовини, що легко проникають через мембрани в ендоплазматичний ретикулум і зв'язуються з одним із цитохромів системи Р446 — Р445 (найчастіше по першому дослідженому ферменті цієї системи вказують тільки цитотохром Р450). Ці цитохроми є первинними компонентами окисної ферментної системи. Швидкість біотрансформації препаратів системою оксидаз змішаного типу визначається концентрацією цитохрому «про, кількістю різних форм цитохрому Р450 і їх спорідненість до субстрату, концентрацією цитохром-с-редуктази і швидкістю відновлення комплексу «препарат - цитохром Р450». Швидкість біотрансформації може залежати і від конкурування ендогенних і екзогенних субстратів.

Подальше окислювання лікарських препаратів відбувається під впливом оксидази і редуктази при обов'язковій участі NADP і молекулярного кисню. Неспецифічні оксидази каталізують процеси дезамінування первинних і вторинних амінів, гідроксилювання бічних ланцюгів і ароматичних кілець гетероциклічних сполук, утворення сульфоксидів і деалкілювання. Кон'югація ЛЗ із глюкуроновою кислотою також здійснюється під впливом мікросомальних ферментів. Це один з основних шляхів біотрансформації карбоновтих кислот, спиртів, фенолів. Шляхом кон'югації з організму виводяться естрогени, глюкокортикоїди, прогестерон, алкалоїди опію й інші наркотичні анальгетики, амідопірин, саліцилати, барбітурати, антибіотики і багато інших речовин.

Лікарські засоби можуть як підвищувати, так і знижувати активність мікросомальних ферментів. Існує велика група речовин, що включаються в печінковий метаболізм, що активують, пригнічують і навіть руйнують цитохром Р450. До їхнього числа відносяться ксикаїн, совкаїн, бенкаїн, індерал, віскен, ералдин і т.д. Більш значною є група речовин, що індукують синтез ферментативних білків печінки, очевидно, за участю NADP, Ш-цитохром Р450-редуктази, цитохрому Р420, N- і О-демилаз мікросом, іонів Mg2+, Ca2+, Мп2+. Це гексобарбітал, фенобарбітал, пентобарбітал, фенілбутазон, кофеїн, етанол, нікотин, бутадієн, нейролептики, амідопірин, хлорциклізин, димедрол, мепробамат, трициклічні антидепресанти, бензонал, хінін, кордіамін, багато хлорвмісних пестицидів. Показано, що в активації даними речовинами ферментів печінки бере участь глюкуронілтрансфераза. При цьому зростає синтез РНК і мікросомальних білків. Індуктори підсилюють не тільки метаболізм ЛЗ в печінці, але і їхнє виведення з жовчю. Причому прискорюється метаболізм лікарських препаратів, що вводяться разом з ними, і самих індукторів.

Немікросомальная біотрансформація Хоча немікросомальні ферменти беруть участь у біотрансформаці невеликого числа ЛЗ, вони, однак, відіграють важливу роль у метаболізмі. Усі види кон'югації, крім глюкуронідної, відновлення і гідролізу лікарських препаратів, каталізуються немікросомальними ферментами. Такі реакції вносять вклад у біотрансформацію ряду розповсюджених лікарських засобів, у тому числі ацетилсаліцилової кислоти і сульфаніламідів. Немікросомальна біотрансформація препаратів відбувається головним чином у печінці, однак вона здійснюється також у плазмі крові й в інших тканинах.

При пероральному застосуванні ЛЗ, що всмоктуються слизовою оболонкою кишечнику, надходять спочатку в портальну систему, а лише потім — у системний кровоток. Інтенсивні і численні реакції метаболізму протікають вже в стінці кишечника (майже усі відомі синтетичні і несинтетичні реакції). Наприклад, ізадрин піддається кон'югації із сульфатами, гідралазин — ацетилуванню. Деякі ЛЗ метаболізуються неспецифічними ферментами (пеніциліни, аміназин) чи бактеріями кишечника (метотрексат, леводопа), що може мати велике практичне значення. Так, у деяких хворих абсорбція аміназину знижена до мінімуму внаслідок значного його метаболізму в кишечнику. Основні процеси біотрансформації відбуваються в печінці.

Метаболізм ЛЗ до поступлення в системний кровоток при проходженні через стінку ЖКТ і печінку називають «ефектом першого проходження». Ступінь метаболізму лікарських засобів при першому проходженні визначається метаболічною ємністю ферментів для даного препарату, швидкістю метаболічних реакцій і абсорбції. Якщо ЛЗ застосовують перорально в невеликій дозі, а ємність ферментів і швидкість метаболізму його значні, то велика частина препарату біотрансформується, за рахунок чого знижується його біодоступність. Зі збільшенням дози лікарського засобу ферментативні системи, що беруть участь у метаболізмі першого проходження, можуть насичуватися, і біодоступність препарату збільшується.
 ліка...")
Виведення ЛЗ з організму Розрізняють кілька шляхів виведення (екскреції) лікарських засобів і їхніх метаболітів з організму. До основного відносяться виведення з сечею і калом, менше значення має виведення з повітрям, потом, слиною і слізною рідиною.

Виведення із сечею. Для оцінки швидкості виведення ЛЗ із сечею визначають його нирковий кліренс. Лікарські препарати виводяться із сечею шляхом клубочкової фільтрації і канальцевої секреції. Велике значення має також їхній реабсорбція в канальцях нирок. Кров, що попадає в нирки, фільтрується в клубочках. При цьому ЛЗ проникають через стінку капілярів у просвіт канальців. Фільтрується тільки та частина препарату, що знаходиться у вільному стані. При проходженні через канальці частина ЛЗ реабсорбується і повертається в плазму крові. Багато ЛЗ активно секретуються з капілярів і перитубулярної рідини в просвіт канальців. При нирковій недостатності клубочкова фільтрація знижується, і виведення різних препаратів порушується, що приводить до збільшення їхньої концентрації в крові. Дозу препаратів, що виводяться із сечею, при прогресуванні уремії варто знизити. Канальцева секреція органічних кислот може бути блокована пробенецидом, що приводить до збільшення періоду їхнього напіввиведення; рН сечі впливає на виведення нирками деяких слабких кислот і основ. Перші швидше виводяться при лужній реакції сечі, а другі — при кислій.

Виведення з жовчю З печінки ЛЗ у виді чи метаболітів у незміненому виді чи пасивно за допомогою активних транспортних систем надходять у жовч. Надалі лікарські препарати чи їхні метаболіти виводяться з організму з калом. Під впливом ферментів ШКТ чи бактеріальної мікрофлори вони можуть перетворюватися в інші сполуки, що реабсорбуються і знову доставляються в печінку, де перетерплюють новий цикл метаболічних перетворень. Подібний цикл зветься ентерогепатичною циркуляцією. На виведення лікарських засобів з жовчю впливають молекулярна маса сполуки, її хімічна природа, стан гепатоцитів і жовчовивідних шляхів, інтенсивність зв'язування препаратів із клітками печінки. Печінковий кліренс препаратів можна визначити при дослідженні дуоденального вмісту, отриманого за допомогою зонда. Ступінь виведення ЛЗ із жовчю особливо важливо враховувати при лікуванні хворих з печінковою недостатністю, а також із запальними захворюваннями жовчних шляхів.
 - основна стру...")
Будова і функції клітини і біологічних мембран Клітина (cytus) - основна структурно-функціональнальна одиниця, що визначає будову, життєдіяльність, розвиток і розмноження тваринних і рослинних організмів за винятком вірусів. Клітини можуть існувати як самостійні організми (бактерії, одноклітинні водорості, гриби, найпростіші) чи утворюють тканини багатоклітинних організмів. Наука про клітину - цитологія - вивчає питання цитоморфології, цитофізіології і цитопатології. У залежності від рівня клітинної організації розрізняють два типи клітин: доядерні (прокаріоти) і ядерні (еукаріоти). Синьо-зелені водорості і бактерії складаються з прокаріотичних клітин, а рослинні і тваринні організми складаються з еукаріотичних клітин.

Еукаріотична клітина складається з трьох основних компартментів: плазматична мембрана, ядро і цитоплазма. Плазматична мембрана (плазмолема) оболонка покриваюча клітину і являє собою комплекс ліпідних і білкових молекул. Ліпідні молекули утворять безупинний подвійний шар товщиною 6-10 нм (ліпідний бішар), у який занурені молекули структурних, транспортних, рецепторних і інших білків. Ліпідний бішар визначає структурні особливості мембрани, а білки - більшість її функцій.
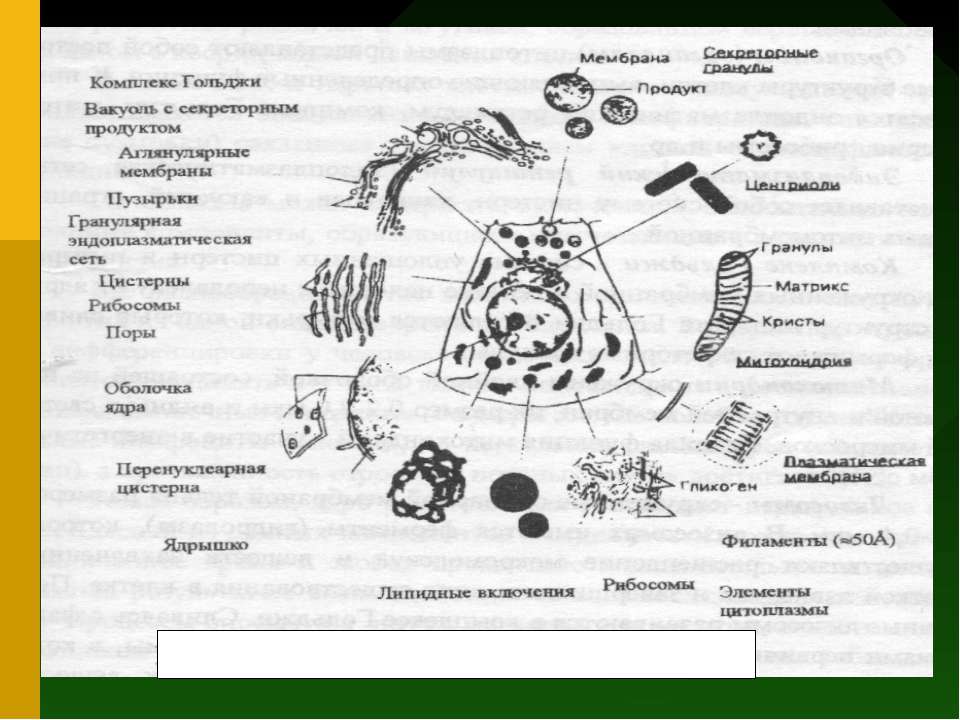
 цитоплазми являють собою постійні структури клітини, що ...")
Органоїди (органели) цитоплазми являють собою постійні структури клітини, що виконують визначені функції. До них відносяться ендоплазматичний ретикулум, комплекс Гольджі, мітохондрії, рибосоми й ін. Ендоплазматичний ретикулум (ендоплазматична мережа) являє собою систему цистерн, канальців і вакуолей, обмежених цитомембраною. Комплекс Гольджі - система сплощених цистерн і пухирців, оточених мембранної і звичайно знаходиться неподалік від ядра. Від структур апарату Гольджі відокремлюються пухирці, що зливаючи, формують секреторні гранули.

Мітохондрії оточені подвійною оболонкою, що складається з зовнішньої і внутрішньої мембран, їхній розмір 0,2-2,0 мкм і видні у світловий мікроскоп. Головна функція мітохондрій - участь в енергетичному обміні. Лізосоми - оточені одинарною мембраною тільця розміром 0,2-0,4 мкм. У лізосомах є ферменти (гідролази), що здійснюють розщеплення макромолекул і речовин захоплених клітиною ззовні, так і тих, що завершили цикл існування в клітині. Первинні лізосоми розвиваються в комплексі Гольджі. Зливаючи з фагосомами первинні лізосоми утворять вторинні лізосоми, у яких відбувається переварювання і засвоєння захоплених речовин. Частина лізосом (аутолізосоми, цитолізосоми) переварюють структури самої клітини, що відмирає. Лізосоми з залишками неперетравлених речовин називають телолізосомами.

Рибосоми - немембранні органоїди, що містяться у всіх живих клітинах, це універсальний апарат синтезу білкових молекул. Вони складаються з білка і РНК і мають розміри 25 20 20 нм. Мікротільця - загальна назва оточених мембраною пухирців діаметром 0,1-1,5 мкм. У них знаходяться ферменти, що каталізують різні окисні реакції. Так, ферменти пероксисом каталізують утворення і руйнування H2О2, що використовується в ряді метаболічних циклів. Клітинний центр (центросома) складається з пари центріолей (диплосома), оточених тонковолокнистою зоною (центросфера). Кожна центріоль має форму циліндра розміром 0,3-0,5 х 1,5 мкм. Це органоїд - центр організації мікротрубочок цитоплазми і зв'язаний з розвитком рісничок і джгутиків, утворенням веретена розподілу й у ціпом з координацією руху клітини.

Біологічні мембрани виконують наступні функції: - бар'єрна; - транспорт речовин; - генерація біоелектричних потенціалів; - процеси трансформації і запасання енергії; - метаболічні функції клітинна рецепція і міжклітинні взаємодії.

Основниі структури еукаріотичної клітини: ядро, занурене в цитоплазму, мембранна система, органоїди, спеціалізовані структури. Ядро складається з каріоплазми (нуклеоплазми), одного чи декількох ядерець і ядерної оболонки. Каріоплазма містить усю хромосомну ДНК клітини, що з білками утворить нитки хроматину - нуклеосоми. У каріоплазмі між нитками хроматину знаходяться кислі білки, багато хто з який беруть участь у регуляції матричної діяльності ділянок ДНК (генів), гранули рибонуклеопротеїнів (РНП), різні ферменти й ін. Ядро оточене ядерною оболонкою, що бере участь в обміні речовин між ядром і цитоплазмою. У цитоплазмі розрізняють: гіалоплазму, мембранну систему, органоїди різні включення. Гіалоплазма - складна біоколоїдна система, що поєднує всі структури клітини і служить середовищем для їхньої взаємодії. Важливу роль у цих процесах відіграють розчинені в цитоплазмі ферменти й АТФ. Крім того, гіалоплазма має тривимірну мережу білкових волоконець. Ряд допоміжних білків зв'язує ці волоконця в каркас - цитоскелет.
Схожі презентації











Категорії