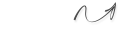Презентація на тему:
Біоорганічна хімія як наука
Завантажити презентацію













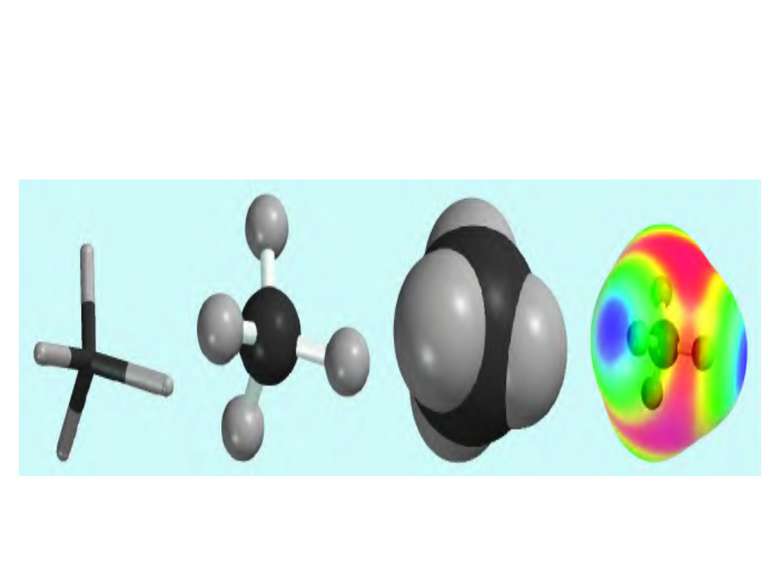





. Правила сучасної номенклатури були розроблен...")









 координація (донорно-акцепторний механізм) – один з атомів є донором елект...")

 – це різновидність ковалентног...")




 відбиває взаємний вплив безпосередн...")
 – це міна розподілу електронн...")

Біоорганічна хімія як наука
Завантажити презентаціюПрезентація по слайдам:

Біоорганічна хімія як наука. Класифікація, номенклатура та реакційна здатність біоорганічних сполук

1. Предмет та значення органічної хімії. Органічна хімія – це хімія атома Карбону та всіх елементів періодичної системи. Органічні речовини відомі людству з давніх-давен. Людство використовувало такі органічні речовини як оцтова кислота, винний спирт, жири, мила, барвники. Етапи становлення органічної хімії. Основні положення теорії хімічної будови органічних сполук О.М.Бутлерова та її значення для розвитку органічної хімії. В історії розвитку хімії для органічної хімії відведено три етапи. Період від першого знайомства людини з органічними речовинами до кінця XVІІІ століття вважається емпіричним. Основний підсумок цього періоду – люди усвідомили значення елементного аналізу і встановлення атомних і молекулярних мас.

Органічна хімія народилась із світу живих організмів. В хімії довгий період часу вважали, що органічні речовини можуть утворюватись тільки в живих організмах під дією “життєвої сили”. Ця теорія довший час існувала і підтримувалась рядом вчених (Берцеліус) і одержала назву “теорії віталізму”. Це вчення сильно гальмувало розвиток органічної хімії, тому що пропагувало ідею про непізнанність світу. До 60-х років XІX століття продовжувався аналітичний період. Він позначався тим, що з кінця першої чверті XІX століття було зроблено ряд перспективних відкриттів, які нанесли нищівного удару по віталістичній теорії. Першим у цій низці був учень Берцеліуса, німецький хімік Велер. Він здійснив ряд відкритті: 1824 р. – синтез щавлевої кислоти з диціану: (CN)2 НООС – СООН. 1828 р. – синтез сечовини з ціановокислого амонію NH4CNO NH2 – C – NH2 О 1842 р. – рос. вчений М.М.Зінін – синтез аніліну з нітробензолу. 1854-1862 – франц. вчений Бертло – синтез оцтової кислоти, ацетилену з С і Н. 1861 р. – О.М.Бутлеров – синтез цукристої речовини.

Накопичений експериментальний матеріал вимагав систематизації, узагальнення. В цей період виникають декілька теорій, серед яких потрібно відзначити теорії типів і радикалів, які виникли одна з другої і розвинулись в постійному протиборстві. Ці теорії відіграли позитивну роль, т.я. сприяли ознайомленню з основними хімічними речовинами. Але в них було закладене те, що вивчити внутрішню будову речовин неможливо (Берцеліус, Велер, Лібіх, Кольбе, Жерар і ін.). Ні одна з цих теорій не могла пояснити і узагальнити дослідний матеріал. В 60-ті роки ці теорії зайшли в тупік. Потрібна була нова теорія. Передбачення її вже були: 1) затверджувалось атомно-молекулярне вчення, створювались поняття “атом”, “молекула як хімічна частинка”; 2) 1857 р. Август Кекуле (нім.) і Купер (шотл.) заклали початки теорії валентності. Вони висловили думку, що вуглець 4-х валентний і може утворювати ланцюги. Але під впливом ідей попередників, вони не змогли відкрити нову теорію, не бачили взаємозв’язку між будовою і властивостями.

Накопичений матеріал вимагав систематизації, узагальнення. Таку теорію розробив учень Зініна Олександр Михайлович Бутлеров (1828-1886). Він ще в 1860 році почав розробляти ідеї теорії хімічної будови речовин. На одній з лекцій в Казанському університеті він висловив свої міркування. Свої положення він базував на дослідженнях попередників. На початку XІX століття при дослідженні органічних сполук хіміки звернули увагу на те, що в ряді хімічних перетворень окремі групи атомів у незмінному вигляді переходять з однієї в іншу ( 1815 р. Ж. Гей-Люссак, 1832 р. Ф.Велер, Ю.Лібіх). Ф.Велер, Ю.Лібіх і Й.Берцеліус створили першу теорію – теорію радикалів. До радикалів вони віднесли групи атомів, що переходять від сполуки до сполуки. Тому вони висловили думку, що органічні речовини складаються з радикалів. У 1833-1834 р.р. Ж.Дюма дослідив, що радикали здатні змінюватись. Це було ударом по теорії радикалів. У 40-х роках XІX століття з’явилась теорія типів (Ш. Жерар). Органічні речовини розглядались як продукти заміщення атомів гідрогену на різні групи. Але ці теорії були неспроможні пояснити явища, які спостерігались і були характерними для органічних сполук.

О.М.Бутлеров висловив свої міркування у 1861 р. на з’їзді природодослідників у м. Карлруе. Він піддав критиці попередні теорії і вказав на їх недоліки. Він дав визначення поняттю “хімічна будова”. За висловом О.М.Бутлерова – це послідовність чергування атомів у молекулі. О.М.Бутлеров писав: “Химическая натура сложной частицы определяется натурой елементарных составных частей, количеством их и химическим строением, а также “химическое значение атомов одного и того же элемента в молекуле обусловлено природой других атомов, соединенных с ним прямо или посредственно”. Постулювання залежності властивостей речовин від хімічної будови відрізняє теорію О.М.Бутлерова від попередніх теорій.

Положення теорії: 1. Атоми, що входять до складу молекули органічних сполук, зв’язані між собою в суворо визначеному порядку, згідно з їх валентністю. Послідовність зв’язування атомів у молекулі називається хімічною будовою. 2. Властивості речовини залежать не тільки від того, які атоми і в якій кількості входять до складу її молекули, але й від того, в якій послідовності вони зв’язані між собою, тобто від хімічної будови молекули. 3. Атоми або групи атомів, які утворюють молекулу, як зв’язані безпосередньо, так і зв’язані через інші атоми, взаємно впливають одні на других, від чого залежить реакційна здатність молекули. 4. Вивчаючи реакційну здатність речовини, можна встановити її будову, і навпаки, за будовою речовини можна судити про її властивості. СКЛАД – БУДОВА – ВЛАСТИВОСТІ Сучасне трактування: Властивості складних речовин залежать від якісного і кількісного складу і від хімічної будови, а також від взаємного впливу атомів в молекулі.

ЗЗначення теорії будови органічних сполук. 1. Зв’язок між будовою і властивостями стверджує пізнаваність органічних речовин і відкриває широку дорогу до вивчення синтезу і структури речовин. Дійсно, з основного положення теорії випливає, що вивчення властивостей речовин дає можливість знати будову і навпаки. 2. Звідси ж випливає пояснення питанню ізомерії (термін був введений в 1830 р. Берцеліусом), а саме, що молекули з однаковим якісним і кількісним складом володіють різними властивостями, так як мають різну будову. 11863 р. – Бутлеров одержав третбутиловий спирт, потім його гомологи, що підтвердило явище ізомерії.

1864 р. – передбачив існування ізомерів в насиченому ряду. 1866 р. – синтезував ізобутан, ізобутилен з третбутилового спирту. 3. Важливою частиною теорії Бутлерова є вчення про взаємний вплив атомів в молекулі. Це вчення було підтверджено учнями Бутлерова – Марковніковим, Зайцевим, Поповим. Теорія хімічної будови вибудувала в струнку систему весь відомий дослідний матеріал. Наступні 100 років тільки підтвердили вірність теорії.

Створення теорії сприяло бурхливому розвитку органічної хімії і дало початок новому структурному періоду, який пов’язаний з синтезом і визначенням структури більшості органічних сполук за допомогою хімічних і фізико-хімічних методів. Цей період в XX столітті характеризується застосуванням автоматизації і комп’ютеризації як в науковій, так і в виробничій сферах. В науковій сфері теорія хімічної будови в 70-х роках збагатилась стереохімічними уявленнями, засновниками якої були гол. вчений Вант-Гофф і франц. Ле Бель. Вони незалежно один від другого опублікували роботи, в яких говорили про тетраедричну будову атома вуглецю в насичених вуглеводнях і пояснювали тим існування оптичних ізомерів. З відкриттям електрона і розвитком уявлень про будову атомів і стану електрона в атомі, поглиблюється і теорія хімічної будови (початок XX століття). У виробничій сфері теорія хімічної будови виявилась тим поштовхом до розвитку, оскільки відкрилась широка можливість до створення нових галузей виробництва.

Сучасна тенденція в розвитку хімічних виробництв заключається в розширенні біохімічних методів (ферментативних) і мікробіологічних (штами мікроорганізмів), за допомогою яких синтезовані інтерферон, інсулін, соматотропін та ін. Досягнення генної інженерії відкривають надзвичайно великі перспективи розвитку народного господарства. Незважаючи на відсутність принципіальної різниці між органічними і неорганічними речовинами, органічна хімія розвинулась як самостійна наука. Цьому сприяло дві особливості: 1) відмінність властивостей органічних і неорганічних речовин: горючість, нестійкість до дії агресивного середовища (кислоти, луги, окислювачі) (досліди з одними і другими речовинами); 2) складаючись з невеликої кількості хімічних елементів (C, H, O, N, Нal, S, P) органічні сполуки нараховують вже понад 14 млн. речовин. Причина відмінностей – переважна кількість сполук з ковалентним зв’язком і здатність атомів Карбону утворювати ланцюги.

3. Способи зображення органічних молекул .


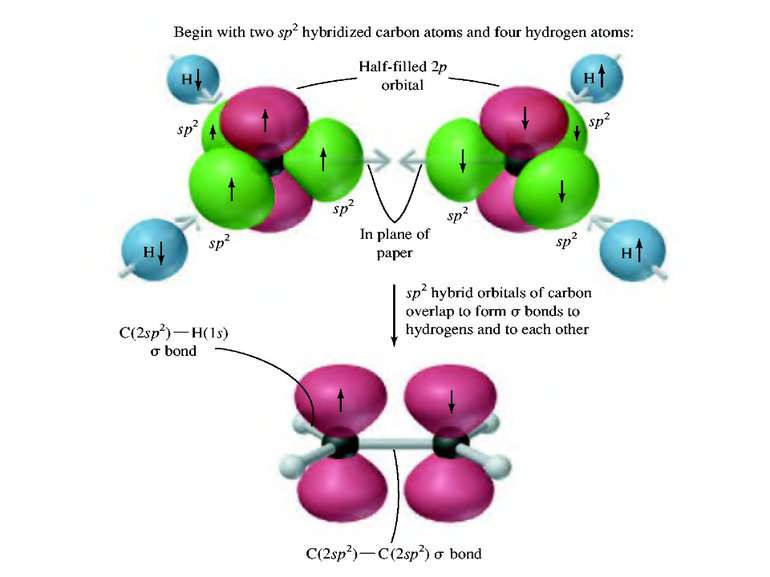
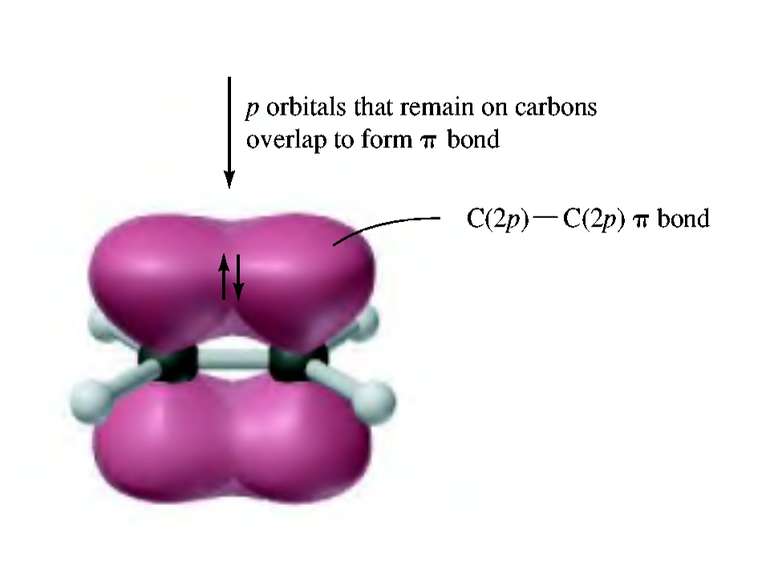
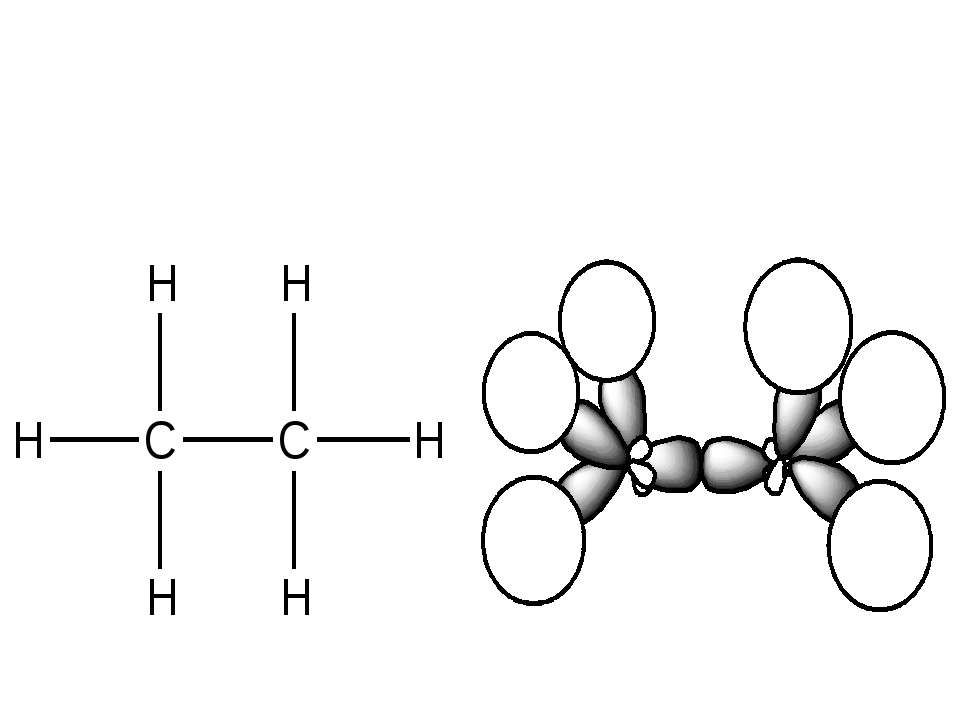
Гібридизація атомних орбіталей: sp-, sp2-, sp3- Atom orbital is the space where the atom can be. There are s–, p– and d–atom orbitals. s-орбіталь p-орбіталь d-орбіталь

sp–гібридизація атомних орбіталей: s-орбіталь p-орбіталь sp-гібридизована орбіталь

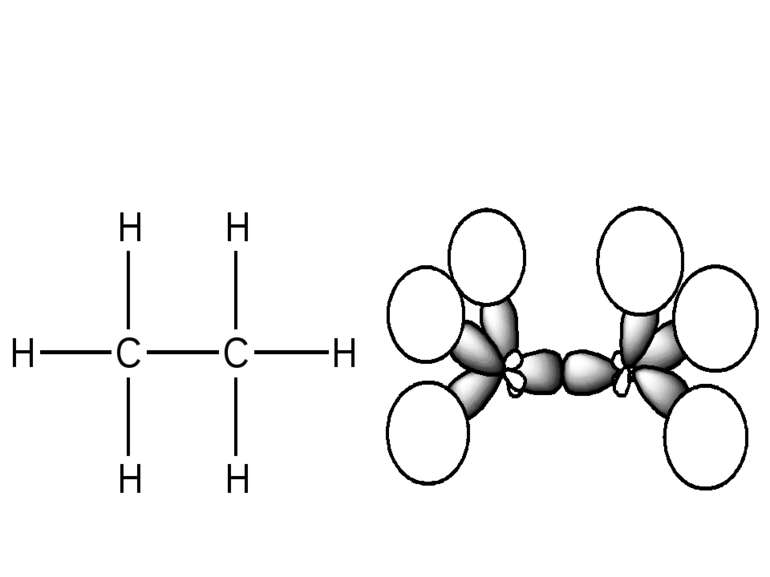
sp2–hybridization of atom orbitals. This hybridization is formed when s–orbital joined to 2 p–orbitals. s-орбіталь p-орбіталі sp2-гібридизовані орбіталі

sp3–hybridization of atom orbitals. This hybridization is formed when s–orbital joined to 3 p–orbitals. s-орбіталь p-орбіталі sp3-гібридизовані орбіталі
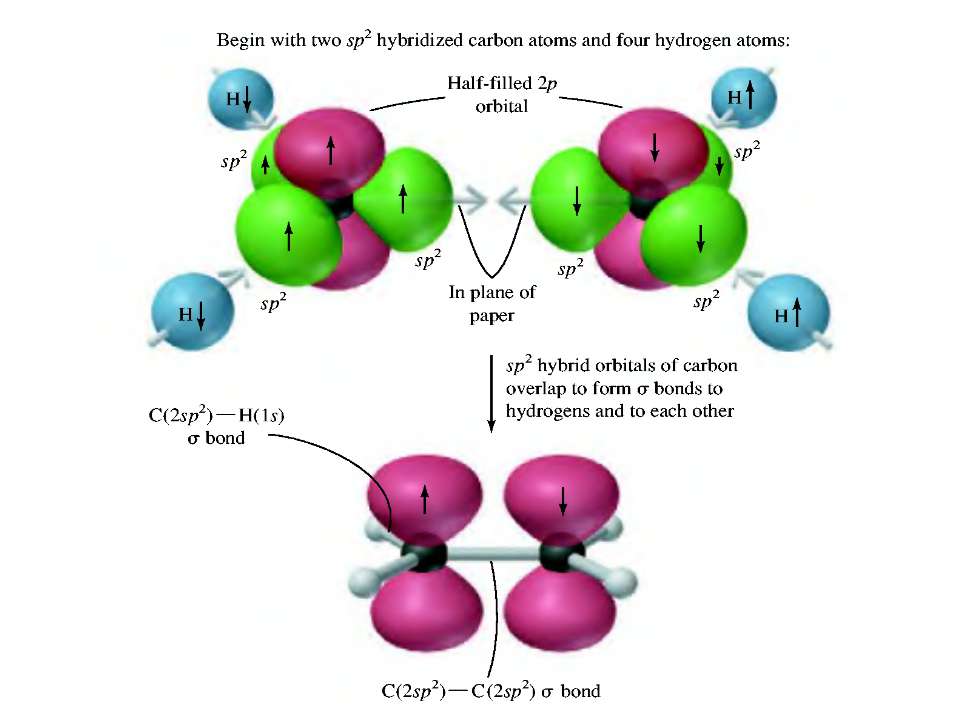
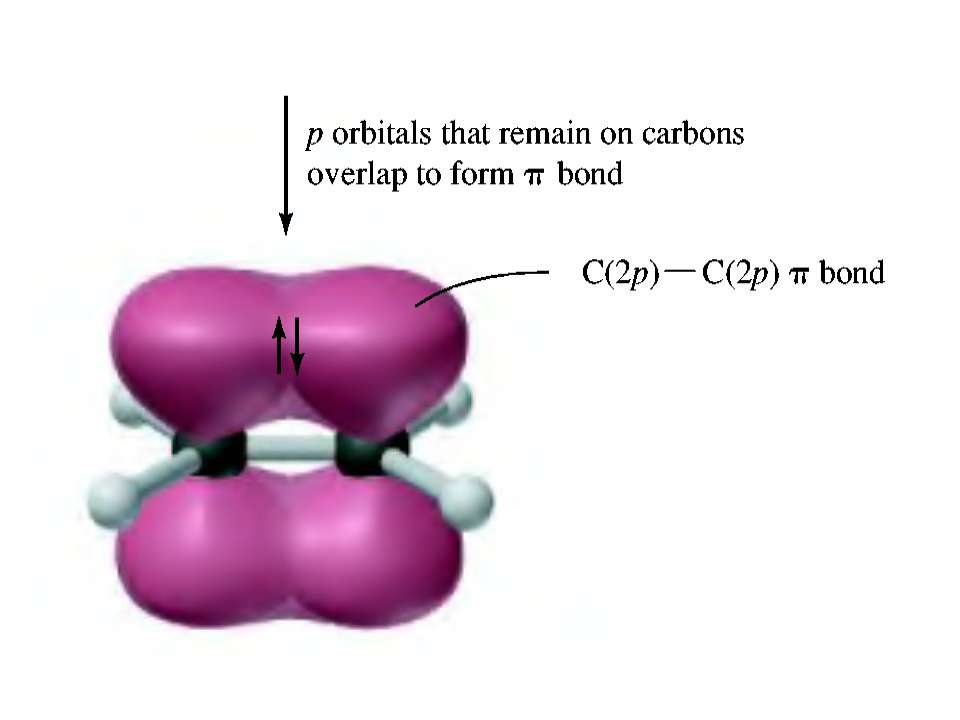

4. Номенклатурні системи в органічній хімії – тривіальна, раціональна та міжнародна (IUPAC) (радикально-функціональна і замісникові). Тривіальна (історична) номенклатура пов’язана із процесом одержання речовин (пірогалол – продукт піролізу галової кислоти), джерела походження, з якого одержували (мурашина кислота) та інше. Тривіальні назви сполук широко застосовують у хімії природних і гетероциклічних сполук (цитраль, гераніол, тіофен, пірол, хінолін та ін.). В основі раціональної номенклатури використовується принцип поділу органічних сполук на гомологічні ряди: усі речовини в певному гомологічному ряді розглядаються як похідні найпростішого представника даного ряду – першого або іноді другого. Зокрема, в алканів – метану, в алкенів – етилену і т. п.
. Правила сучасної номенклатури були розроблен...")
Міжнародна номенклатура (IUPAC). Правила сучасної номенклатури були розроблені у 1957 році на ХІХ конгресі Міжнародного союзу теоретичної і прикладної хімії (International Union of Pure and Applied Chemistry – IUPAC). Розглянемо замісникові та радикально-функціональну номенклатуру ІЮПАК. Замісникові номенклатура. Правила номенклатури. 1. При утворенні назв за замісниковою номенклатурою органічні сполуки розглядають як похідні найпростіших вуглеводнів, у молекулі яких один або декілька атомів Гідрогену замішені на інші атоми або групи атомів, які називаються замісниками. 2. Визначають, які функціональні групи входять до складу сполуки і вибирають серед них старшу, якщо вона є: – COOH > –SO3H > –COOR > –C(O)Cl > –C(O)NH2 > –C N > –C(O)H > – >C=O > –OH > –SH > –NH2 > –Hal (F, Cl, Br, I). 3. Визначають родоначальну структуру молекули. Це є структурний фрагмент молекули, що лежить в основі назви. В ациклічних сполуках – це головний вуглецевий ланцюг, у карбоциклічних і гетероциклічних – цикл.

За головний вуглецевий ланцюг обирають той, котрий містить максимальне (у порядку убиваючої значимості) число: 1) функціональних груп; 2) кратних зв’язків; 3) атомів Карбону; 4) замісників. Замісником називають будь-який атом або групу атомів, котрі не входять до родоначальної структури. Поняття “замісник” включає у себе функціональну групу і радикал. Радикал – це залишок молекули вуглеводню, що утворюється в результаті видалення одного або декількох атомів Гідрогену. Утворену вільну валентність позначають рискою. За кількістю вільних валентностей розрізняють одно- дво- і тривалентні радикали: В залежності від природи атома Карбону розрізняють первинні, вторинні і третинні радикали. (А чи можуть бути четвертинні радикали?)

4. Визначивши родоначальну структуру, нумерують її атоми таким чином, щоб старша дістала, по можливості, найменший номер. Якщо старшої групи немає, перевагу надають кратним зв’язкам, а при їх відсутності – замісникам. 5. Складають назву сполуки, дотримуючись такої послідовності: - алфавітний порядок функціональних груп, окрім старшої, та вуглеводневих радикалів (префікс); - назва родоначальної структури (корінь); - кратні зв’язки і старшу функціональну групу (суфікс). = зв’язок – “ен”, зв’язок – “ин(ін)” Кількість замісників: ди – (два), три – (три), тетра – (чотири), пента – п’ять. Радикально-функціональна номенклатура. В основі цих назв лежить назва функціонального класу (спирт, етер, кетон і ін.), якому передують назви вуглеводневих радикалів, наприклад: алілхлорид, діетиловий етер, диметилкетон, пропіловий спирт.

У випадку наявності в сполуці декількох функціональних груп, то саме старша функціональна група визначає приналежність до класу, а замісники позначають літерами грецького алфавіту , , , і т. д., причому літерою позначається перший атом від функціональної групи, а для назви родоначальної структури використовують тривіальну назву: аланін (тривіальна назва) -амінопропіонова кислота (радикально-функціональна) 2-амінопропанова кислота (замісникова ІЮПАК)

6. Структурна ізомерія органічних сполук: ізомерія вуглецевого скелету, ізомерія положення, ізомерія функціональної групи. Правила виведення ізомерів. Поняття про таутомерію.

ССтатична структурна ізомерія – це ізомерія речовин, яка пов’язана з певною послідовністю сполучення атомів між собою в молекулах і при якій ізомери не перетворюються самовільно один в одного. ППравила виведення ізомерів 1. Перший ізомер має в ланцюгу найбільшу кількість атомів Карбону. 2. Другий ізомер має в ланцюгу на один атом Карбону менше. Пам’ятайте, алкільні радикали не можна ставити біля першого і останнього атомів Карбону основного ланцюга. Алкільні радикали розміщують тільки біля нерівноцінних атомів Карбону. 3. При заданій кількості атомів Карбону проводять переміщення функціональних груп. 4.

Надалі знову зменшують кількість атомів Карбону в основному ланцюгу на один. Пам’ятайте, радикали етил і інші не можна ставити біля другого і передостаннього атомів Карбону основного ланцюга. Різні ізомери повинні мати різну хімічну назву (при цьому слід дотримуватись правил замісникової номенклатури).

ТТаутомерія – динамічна структурна ізомерія, яка пов’язана з оборотним перенесенням атомів у вигляді катіона або аніона від однієї молекули до іншої. Найчастіше відбувається перенесення протона Н+ і тому таку таутомерію називають прототропною. До неї належить таутомерія, що пов’язана з підвищеною рухливістю атома Гідрогену в -положенні до електроноакцепторних груп. ВВиди таутомерії: 11) Кето-енольна 2) Нітро-аци-нітро таутомерія 33) Лактим-лактамна таутомерія 4) Азольна таутомерія 55) Імін-енамінна таутомерія 6) Нітрозо-оксимна таутомерія

Одне з головних питань – питання про хімічний зв’язок. Це питання було розв’язане тільки після відкриття електрону. На початку XX ст., коли були досягнуті успіхи в питанні будови електронних оболонок атомів, коли стало відомо, що властивості атомів визначаються переважно електронами останнього енергетичного рівня, питання про хімічний зв’язок набрало ваги. Розробка електронних уявлень в органічній хімії (Штарк, Коссель, Льюїс, Ленгмюр) приводить до виникнення “октетної теорії”, або “теорії електронних пар” (1916-1917 р.) для практикування природи хімічного зв’язку. Автори звернули увагу на стабільність конфігурації з 8 електронів (інертні гази) і на те, що при утворенні зв’язку атоми намагаються утворити стійкий октет. (формули Льюїса)

У відповідності з октетною теорією утворення октету може проходити двома шляхами, тому є два види хімічного зв’язку: гетерополярний і гомеополярний зв’язки. 1. Гетерополярний, електровалентний (Коссель, Ленгмюр) або іонна взаємодія пов’язана з повною віддачею атомами електронів. Атоми перетворюються в іони. 2. Гомеополярний зв’язок, атомний. Стійкі октети створюються шляхом узагальнення електронів. Зв’язок створений спільними електронними парами, називається ковалентним. Виникнення ковалентного зв’язку йде двома механізмами: а) коллігація (обмінний механізм), кожний з атомів дає електрони для спільного користування): СН3 + СН3 СН3 – СН3
 координація (донорно-акцепторний механізм) – один з атомів є донором елект...")
б) координація (донорно-акцепторний механізм) – один з атомів є донором електронів, а другий – акцептором. СН3 + СН3 СН3 – СН3

Ці два методи – це два різних шляхи утворення одного і того ж зв’язку, структура якого не залежить від механізму утворення. По донорно-акцепторному механізму взаємодіють сполуки, які містять в своєму складі атоми N, O, S. Льюїс розглядає подібні реакції як кислотно-основні. Донори – основи, акцептори – кислоти. хлорид метиламонію
 – це різновидність ковалентног...")
Семіполярний зв’язок (біполярний, півполярний) – це різновидність ковалентного зв’язку, утвореного по донорно-акцепторному механізму. Це такий зв’язок, коли ковалентнозв’язані атоми несуть повні протилежні (формальні) заряди. Цей вид зв’язку являє собою комбінацію ковалентного і електровалентного зв’язку. Від звичайного ковалентного зв’язку відрізняється тим, що атоми несуть заряди; від електронновалентного тим, що крім іонної взаємодії атоми зв’язані ще й ковалентно, тому сполуки не дисоціюють у водному розчині на іони.

Водневий зв’язок Водневий зв’язок відноситься до особливого типу донорно-акцепторного зв’язку. Водневий зв’язок виникає у випадку, коли Гідроген зв’язаний ковалентно з сильно електронегативним (ЕН) елементом (F, O, N). Електронна пара при цьому значно зміщена до ЕН атому, а над Гідрогеном виникає +. При наближенні до атома Гідрогену іншого електронегативного атома іншої молекули, виникають сили взаємодії. При цьому ЕН атом виступає в якості донора електронів, а Гідроген – в якості акцептора. Атом Гідрогену займає проміжне положення між двома ЕН атомами, з одним з яких він зв’язаний ковалентно. Водневий зв’язок буває міжмолекулярним і внутрішньомолекулярним.

Утворення міжмолекулярного зв’язку у випадку води, спиртів, приводить до збільшення Тк. Інколи зв’язки дуже міцні, утворюються асоціати: n – нітрофенол (не переганяються з водяним паром) о-нітрофенол (внутрішньо-молекулярні зв’язки) переганяються з водяною парою. Розчинність нижчих спиртів і амінів пов’язана з їх здатністю утворювати водневі зв’язки.

Водневі зв’язки дуже поширені в природі, вони позначаються на властивостях білків, нуклеїнових кислот. Функціональні властивості білків визначаються їх здатністю утворювати водневі зв’язки. Ген-фрагмент ДНК – складна молекула, яка повинна зберегти свою конфігурацію, щоб вірно передавати спадковість. Водневі зв’язки відіграють важливу роль в підтриманні цієї конфігурації. Відомо, що з усіх взаємодій лише водневі зв’язки мають потрібну міцність і направленість, які характеризуються необхідними для підтримання структур молекул.

Деякі властивості ковалентних зв’язків Ковалентний зв’язок характеризується довжиною, енергією, полярністю, поляризованістю, насиченістю і направленістю. Дві останні властивості відсутні при іонній взаємодії. Довжина зв’язку (l) – віддаль між ядрами атомів, що відповідає мінімальній енергії системи з двох ядер. 10А0 – 1нм. Сsp3 – Сsp3 0,154 нм, Сsp3 – Сsp2 0,150 нм Сsp3 – Сsp3 0,146 нм С = С 0,134 нм С С 0,120 нм С – Н 0,111 нм Енергія зв’язку (Е) – є мірою його міцності і визначається як кількість енергії, що виділяється при утворенні зв’язку, або яку потрібно затратити на його розрив. Виражається в кДж/моль 1 ккал = 4,184 кДж. С – С 330 - 360 кДж/моль, С = С 590 - 640 кДж/моль, С С 810 - 840 кДж/моль, С – Н 402 - 455 кДж/моль. Просторова направленість sp3 – гібридизація тетраедрична будова sp2 – гібридизація площинна будова sp – гібридизація лінійна будова
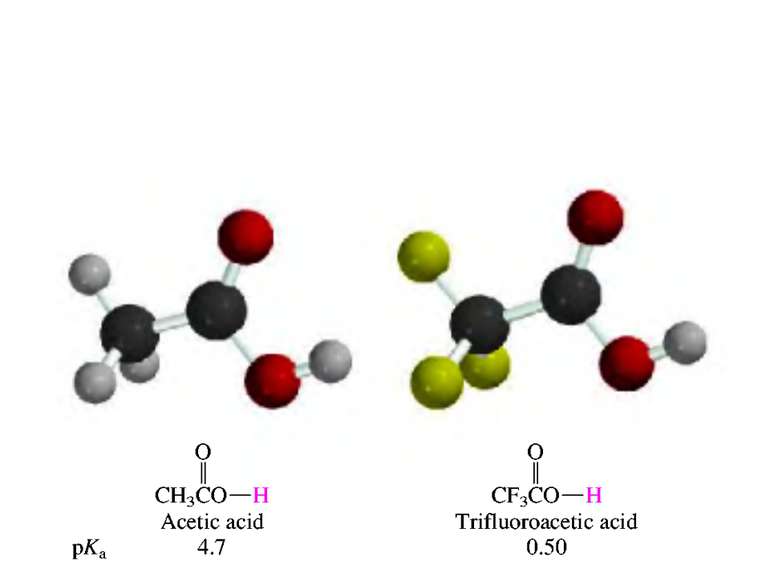
 відбиває взаємний вплив безпосередн...")
Полярність зв’язку (статична поляризація) відбиває взаємний вплив безпосередньо зв’язаних атомів. Це зміщення електронної хмари зв’язку до більш електронегативного атома. Електронегативність – здатність атома або групи атомів відтягувати до себе електронну густину. Електронегативність елементів визначається шкалою Полінга: 4,0 3,5 3,2 3,0 2,8 2,75 2,6 2,5 2,2 F > O > Csp > N, Cl > Br > Csp2 > I > Csp3 > H Чим більша електронегативність атома, тим більш полярний зв’язок. Кількісно полярний зв’язок можна охарактеризувати величиною дипольного моменту ( ). Полярні молекули утворюють диполі. Диполь – система з розділеними центрами електропозитивних і електронегативних зарядів. Одиниці виміру дипольного моменту Д (дебай) або Кулон метр (Кл м). = g l 1 Д = 3,33 10–30 Кл м. Для ковалентних молекул = 0 – 4 Д, для йонних – = 4 – 11 Д. Дипольний момент – величина векторна, показується від + до – . В молекулі дипольний момент являє собою геометричну суму дипольних моментів окремих зв’язків. Тому деякі симетричні молекули не полярні.
 – це міна розподілу електронн...")
Поляризованість зв’язку (динамічна поляризація) – це міна розподілу електронної густини під дією зовнішніх чинників (реагентів, розчинників, каталізаторів). Поляризованість зростає із зменшенням ЕН та із збільшенням радіуса атома і є більш визначальною на реакційну здатність молекули. Чим більше електрони зсунуті в статичній молекулі, тим менше залишається можливостей для їх зсуву під дією зовнішнього поля. Найменш полярна молекула HJ являє собою саму сильну кислоту: у водному середовищі завдяки високій поляризованість легко відщеплює протон. Поляризованість молекул має важливе значення для пояснення поведінки речовин в момент реакції.

Сучасний спектрофотометр для УФ та видимої ділянок спектру

Схожі презентації











Категорії